WGS Somatic (Multi callers)¶
WGSSomaticMultiCallers · A somatic tumor-normal variant-calling WGS pipeline using GATK, VarDict and Strelka2 · 3 contributors · 1 version
This is a genomics pipeline to align sequencing data (Fastq pairs) into BAMs:
- Takes raw sequence data in the FASTQ format;
- align to the reference genome using BWA MEM;
- Marks duplicates using Picard;
- Call the appropriate somatic variant callers (GATK / Strelka / VarDict);
- Outputs the final variants in the VCF format.
Resources
This pipeline has been tested using the HG38 reference set, available on Google Cloud Storage through:
This pipeline expects the assembly references to be as they appear in that storage (“.fai”, “.amb”, “.ann”, “.bwt”, “.pac”, “.sa”, “^.dict”). The known sites (snps_dbsnp, snps_1000gp, known_indels, mills_indels) should be gzipped and tabix indexed.
Quickstart¶
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
| Name | Type | Source | Description |
|---|---|---|---|
| reference | FastaWithIndexes |
|
The reference genome from which to align the reads. This requires a number indexes (can be generated with the ‘IndexFasta’ pipeline This pipeline has been tested using the HG38 reference set.
|
| snps_dbsnp | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| snps_1000gp | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/ |
| known_indels | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| mills_indels | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| gatk_intervals | Array<bed> | None | List of intervals over which to split the GATK variant calling |
| gridss_blacklist | bed | BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist | |
| vardict_intervals | Array<bed> | None | List of intervals over which to split the VarDict variant calling |
| strelka_intervals | Gzipped<bed> | None | An interval for which to restrict the analysis to. |
| gnomad | Gzipped<VCF> | The genome Aggregation Database (gnomAD). This VCF must be compressed and tabix indexed. This is specific for your genome (eg: hg38 / br37) and can usually be found with your reference. For example for HG38, the Broad institute provide the following af-only-gnomad compressed and tabix indexed VCF: https://console.cloud.google.com/storage/browser/gatk-best-practices/somatic-hg38;tab=objects?prefix=af-only | |
| panel_of_normals | Optional<Gzipped<VCF>> |
|
VCF file of sites observed in normal. |
| cutadapt_adapters | File | https://raw.githubusercontent.com/csf-ngs/fastqc/master/Contaminants/contaminant_list.txt |
|
- Generate user and static input files for WGSSomaticMultiCallers:
# user inputs
janis inputs --user WGSSomaticMultiCallers > inputs.yaml
# static inputs
janis inputs --static WGSSomaticMultiCallers > static.yaml
inputs.yaml
normal_inputs: '["normal_R1.fastq.gz", "normal_R2.fastq.gz"]'
normal_name: NA24385_normal
tumor_inputs: '["tumor_R1.fastq.gz", "tumor_R2.fastq.gz"]'
tumor_name: NA24385_tumor
static.yaml
cutadapt_adapters: contaminant_list.txt
gatk_intervals: BRCA1.bed
gnomad: af-only-gnomad.hg38.vcf.gz
gridss_blacklist: gridss_blacklist.bed
known_indels: Homo_sapiens_assembly38.known_indels.vcf.gz
mills_indels: Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
reference: Homo_sapiens_assembly38.fasta
snps_1000gp: 1000G_phase1.snps.high_confidence.hg38.vcf.gz
snps_dbsnp: Homo_sapiens_assembly38.dbsnp138.vcf.gz
strelka_intervals: BRCA1.bed.gz
vardict_intervals: BRCA1.bed
- Run WGSSomaticMultiCallers with:
janis run [...run options] \
--inputs inputs.yaml \
--inputs static.yaml \
WGSSomaticMultiCallers
Outputs¶
| name | type | documentation |
|---|---|---|
| out_normal_fastqc_reports | Array<Array<Zip>> | |
| out_tumor_fastqc_reports | Array<Array<Zip>> | |
| out_normal_performance_summary | csv | A text file of performance summary of NORMAL bam |
| out_tumor_performance_summary | csv | A text file of performance summary of TUMOR bam |
| out_normal_bam | IndexedBam | |
| out_tumor_bam | IndexedBam | |
| out_gridss_assembly | BAM | Assembly returned by GRIDSS |
| out_variants_gridss | VCF | Variants from the GRIDSS variant caller |
| out_variants_gatk | Gzipped<VCF> | Merged variants from the GATK caller |
| out_variants_split | Array<VCF> | Unmerged variants from the GATK caller (by interval) |
| out_variants_vardict_split | Array<VCF> | Unmerged variants from the VarDict caller (by interval) |
| out_variants_vardict | Gzipped<VCF> | Merged variants from the VarDict caller |
| out_variants_strelka | VCF | Variants from the Strelka variant caller |
| out_variants | VCF | Combined variants from GATK, VarDict and Strelka callers |
Workflow¶
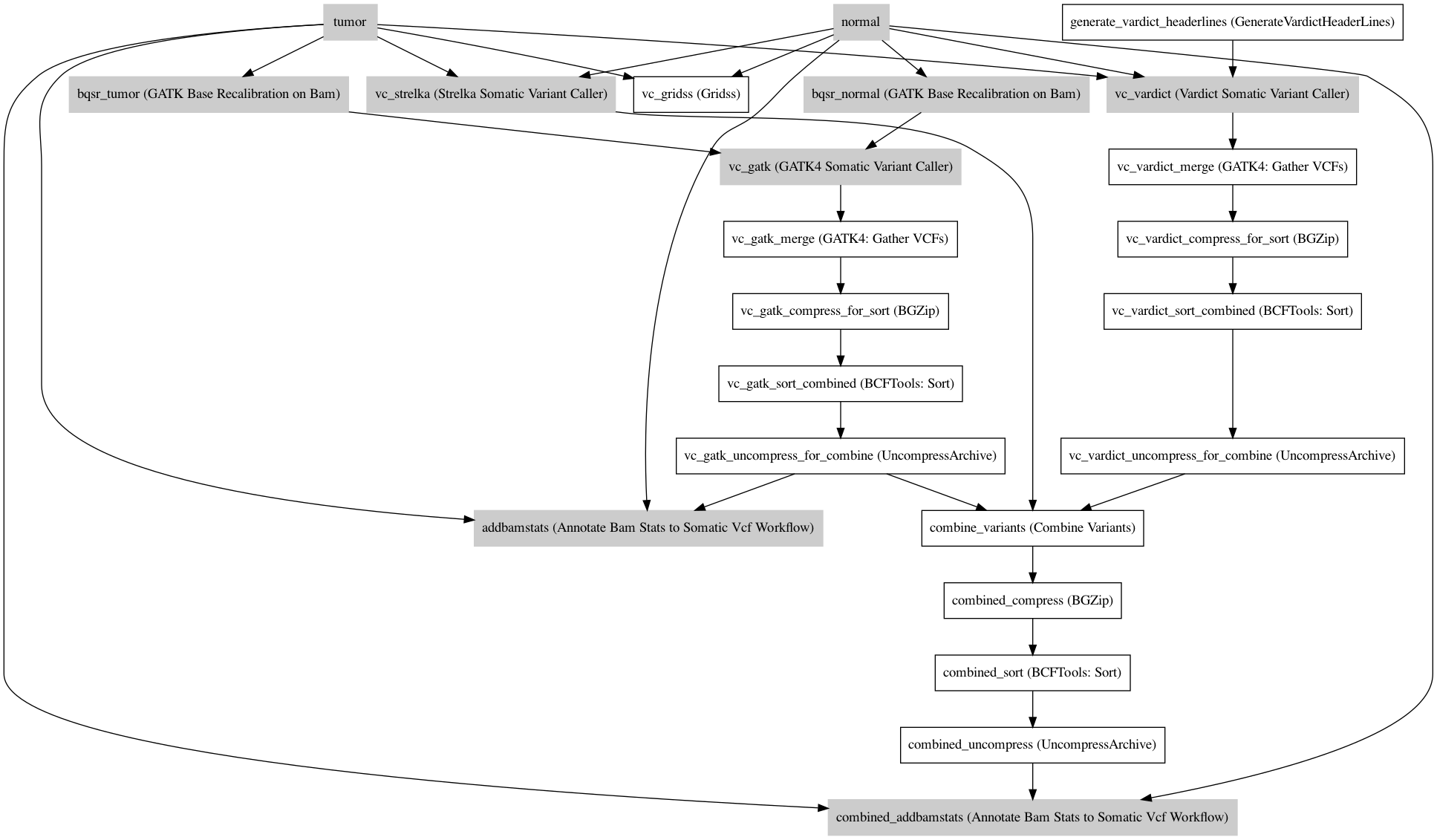
Information¶
| ID: | WGSSomaticMultiCallers |
|---|---|
| Versions: | 1.4.0 |
| Authors: | Michael Franklin, Richard Lupat, Jiaan Yu |
| Citations: | |
| Created: | 2018-12-24 |
| Updated: | 2020-08-19 |
Embedded Tools¶
| Gridss | gridss/v2.6.2 |
| GATK Base Recalibration on Bam | GATKBaseRecalBQSRWorkflow/4.1.3 |
| GATK4 Somatic Variant Caller | GATK4_SomaticVariantCaller/4.1.3.0 |
| GATK4: Gather VCFs | Gatk4GatherVcfs/4.1.3.0 |
| BGZip | bgzip/1.2.1 |
| BCFTools: Sort | bcftoolssort/v1.9 |
| UncompressArchive | UncompressArchive/v1.0.0 |
| Annotate Bam Stats to Somatic Vcf Workflow | AddBamStatsSomatic/v0.1.0 |
| GenerateVardictHeaderLines | GenerateVardictHeaderLines/v0.1.0 |
| Vardict Somatic Variant Caller | vardictSomaticVariantCaller/v0.1.0 |
| Strelka Somatic Variant Caller | strelkaSomaticVariantCaller/v0.1.1 |
| Combine Variants | combinevariants/0.0.8 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| normal_inputs | Array<FastqGzPair> | An array of NORMAL FastqGz pairs. These are aligned separately and merged to create higher depth coverages from multiple sets of reads |
| tumor_inputs | Array<FastqGzPair> | An array of TUMOR FastqGz pairs. These are aligned separately and merged to create higher depth coverages from multiple sets of reads |
| normal_name | String | Sample name for the NORMAL sample from which to generate the readGroupHeaderLine for BwaMem |
| tumor_name | String | Sample name for the TUMOR sample from which to generate the readGroupHeaderLine for BwaMem |
| reference | FastaWithIndexes | The reference genome from which to align the reads. This requires a number indexes (can be generated with the ‘IndexFasta’ pipeline This pipeline has been tested using the HG38 reference set.
|
| snps_dbsnp | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| snps_1000gp | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/ |
| known_indels | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| mills_indels | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| gatk_intervals | Array<bed> | List of intervals over which to split the GATK variant calling |
| gridss_blacklist | bed | BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist |
| vardict_intervals | Array<bed> | List of intervals over which to split the VarDict variant calling |
| strelka_intervals | Gzipped<bed> | An interval for which to restrict the analysis to. |
| gnomad | Gzipped<VCF> | The genome Aggregation Database (gnomAD). This VCF must be compressed and tabix indexed. This is specific for your genome (eg: hg38 / br37) and can usually be found with your reference. For example for HG38, the Broad institute provide the following af-only-gnomad compressed and tabix indexed VCF: https://console.cloud.google.com/storage/browser/gatk-best-practices/somatic-hg38;tab=objects?prefix=af-only |
| cutadapt_adapters | File |
|
| panel_of_normals | Optional<Gzipped<VCF>> | VCF file of sites observed in normal. |
| allele_freq_threshold | Optional<Float> | The threshold for VarDict’s allele frequency, default: 0.05 or 5% |
| combine_variants_type | Optional<String> | germline | somatic |
| combine_variants_columns | Optional<Array<String>> | Columns to keep, seperated by space output vcf (unsorted) |
Workflow Description Language¶
version development
import "tools/somatic_subpipeline.wdl" as S
import "tools/gridss_v2_6_2.wdl" as G
import "tools/GATKBaseRecalBQSRWorkflow_4_1_3.wdl" as G2
import "tools/GATK4_SomaticVariantCaller_4_1_3_0.wdl" as G3
import "tools/Gatk4GatherVcfs_4_1_3_0.wdl" as G4
import "tools/bgzip_1_2_1.wdl" as B
import "tools/bcftoolssort_v1_9.wdl" as B2
import "tools/UncompressArchive_v1_0_0.wdl" as U
import "tools/AddBamStatsSomatic_v0_1_0.wdl" as A
import "tools/GenerateVardictHeaderLines_v0_1_0.wdl" as G5
import "tools/vardictSomaticVariantCaller_v0_1_0.wdl" as V
import "tools/strelkaSomaticVariantCaller_v0_1_1.wdl" as S2
import "tools/combinevariants_0_0_8.wdl" as C
workflow WGSSomaticMultiCallers {
input {
Array[Array[File]] normal_inputs
Array[Array[File]] tumor_inputs
String normal_name
String tumor_name
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
File snps_dbsnp
File snps_dbsnp_tbi
File snps_1000gp
File snps_1000gp_tbi
File known_indels
File known_indels_tbi
File mills_indels
File mills_indels_tbi
Array[File] gatk_intervals
File gridss_blacklist
Array[File] vardict_intervals
File strelka_intervals
File strelka_intervals_tbi
File gnomad
File gnomad_tbi
File? panel_of_normals
File? panel_of_normals_tbi
File cutadapt_adapters
Float? allele_freq_threshold = 0.05
String? combine_variants_type = "somatic"
Array[String]? combine_variants_columns = ["AD", "DP", "GT"]
}
call S.somatic_subpipeline as tumor {
input:
reads=tumor_inputs,
sample_name=tumor_name,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
cutadapt_adapters=cutadapt_adapters,
gatk_intervals=gatk_intervals,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
call S.somatic_subpipeline as normal {
input:
reads=normal_inputs,
sample_name=normal_name,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
cutadapt_adapters=cutadapt_adapters,
gatk_intervals=gatk_intervals,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
call G.gridss as vc_gridss {
input:
bams=[normal.out_bam, tumor.out_bam],
bams_bai=[normal.out_bam_bai, tumor.out_bam_bai],
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
blacklist=gridss_blacklist
}
scatter (g in gatk_intervals) {
call G2.GATKBaseRecalBQSRWorkflow as bqsr_normal {
input:
bam=normal.out_bam,
bam_bai=normal.out_bam_bai,
intervals=g,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
}
scatter (g in gatk_intervals) {
call G2.GATKBaseRecalBQSRWorkflow as bqsr_tumor {
input:
bam=tumor.out_bam,
bam_bai=tumor.out_bam_bai,
intervals=g,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
}
scatter (Q in zip(gatk_intervals, zip(transpose([bqsr_normal.out, bqsr_normal.out_bai]), transpose([bqsr_tumor.out, bqsr_tumor.out_bai])))) {
call G3.GATK4_SomaticVariantCaller as vc_gatk {
input:
normal_bam=Q.right.left[0],
normal_bam_bai=Q.right.left[1],
tumor_bam=Q.right.right[0],
tumor_bam_bai=Q.right.right[1],
normal_name=normal_name,
intervals=Q.left,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
gnomad=gnomad,
gnomad_tbi=gnomad_tbi,
panel_of_normals=panel_of_normals,
panel_of_normals_tbi=panel_of_normals_tbi
}
}
call G4.Gatk4GatherVcfs as vc_gatk_merge {
input:
vcfs=vc_gatk.out
}
call B.bgzip as vc_gatk_compress_for_sort {
input:
file=vc_gatk_merge.out
}
call B2.bcftoolssort as vc_gatk_sort_combined {
input:
vcf=vc_gatk_compress_for_sort.out
}
call U.UncompressArchive as vc_gatk_uncompress_for_combine {
input:
file=vc_gatk_sort_combined.out
}
call A.AddBamStatsSomatic as addbamstats {
input:
normal_id=normal_name,
tumor_id=tumor_name,
normal_bam=normal.out_bam,
normal_bam_bai=normal.out_bam_bai,
tumor_bam=tumor.out_bam,
tumor_bam_bai=tumor.out_bam_bai,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
vcf=vc_gatk_uncompress_for_combine.out
}
call G5.GenerateVardictHeaderLines as generate_vardict_headerlines {
input:
reference=reference,
reference_dict=reference_dict
}
scatter (v in vardict_intervals) {
call V.vardictSomaticVariantCaller as vc_vardict {
input:
normal_bam=normal.out_bam,
normal_bam_bai=normal.out_bam_bai,
tumor_bam=tumor.out_bam,
tumor_bam_bai=tumor.out_bam_bai,
normal_name=normal_name,
tumor_name=tumor_name,
intervals=v,
allele_freq_threshold=select_first([allele_freq_threshold, 0.05]),
header_lines=generate_vardict_headerlines.out,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
}
call G4.Gatk4GatherVcfs as vc_vardict_merge {
input:
vcfs=vc_vardict.out
}
call B.bgzip as vc_vardict_compress_for_sort {
input:
file=vc_vardict_merge.out
}
call B2.bcftoolssort as vc_vardict_sort_combined {
input:
vcf=vc_vardict_compress_for_sort.out
}
call U.UncompressArchive as vc_vardict_uncompress_for_combine {
input:
file=vc_vardict_sort_combined.out
}
call S2.strelkaSomaticVariantCaller as vc_strelka {
input:
normal_bam=normal.out_bam,
normal_bam_bai=normal.out_bam_bai,
tumor_bam=tumor.out_bam,
tumor_bam_bai=tumor.out_bam_bai,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
intervals=strelka_intervals,
intervals_tbi=strelka_intervals_tbi
}
call C.combinevariants as combine_variants {
input:
vcfs=[vc_gatk_uncompress_for_combine.out, vc_strelka.out, vc_vardict_uncompress_for_combine.out],
type=select_first([combine_variants_type, "somatic"]),
columns=select_first([combine_variants_columns, ["AD", "DP", "GT"]]),
normal=normal_name,
tumor=tumor_name
}
call B.bgzip as combined_compress {
input:
file=combine_variants.out
}
call B2.bcftoolssort as combined_sort {
input:
vcf=combined_compress.out
}
call U.UncompressArchive as combined_uncompress {
input:
file=combined_sort.out
}
call A.AddBamStatsSomatic as combined_addbamstats {
input:
normal_id=normal_name,
tumor_id=tumor_name,
normal_bam=normal.out_bam,
normal_bam_bai=normal.out_bam_bai,
tumor_bam=tumor.out_bam,
tumor_bam_bai=tumor.out_bam_bai,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
vcf=combined_uncompress.out
}
output {
Array[Array[File]] out_normal_fastqc_reports = normal.out_fastqc_reports
Array[Array[File]] out_tumor_fastqc_reports = tumor.out_fastqc_reports
File out_normal_performance_summary = normal.out_performance_summary
File out_tumor_performance_summary = tumor.out_performance_summary
File out_normal_bam = normal.out_bam
File out_normal_bam_bai = normal.out_bam_bai
File out_tumor_bam = tumor.out_bam
File out_tumor_bam_bai = tumor.out_bam_bai
File out_gridss_assembly = vc_gridss.assembly
File out_variants_gridss = vc_gridss.out
File out_variants_gatk = vc_gatk_sort_combined.out
Array[File] out_variants_split = vc_gatk.out
Array[File] out_variants_vardict_split = vc_vardict.out
File out_variants_vardict = vc_vardict_sort_combined.out
File out_variants_strelka = vc_strelka.out
File out_variants = addbamstats.out
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: WGS Somatic (Multi callers)
doc: |
This is a genomics pipeline to align sequencing data (Fastq pairs) into BAMs:
- Takes raw sequence data in the FASTQ format;
- align to the reference genome using BWA MEM;
- Marks duplicates using Picard;
- Call the appropriate somatic variant callers (GATK / Strelka / VarDict);
- Outputs the final variants in the VCF format.
**Resources**
This pipeline has been tested using the HG38 reference set, available on Google Cloud Storage through:
- https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
This pipeline expects the assembly references to be as they appear in that storage (".fai", ".amb", ".ann", ".bwt", ".pac", ".sa", "^.dict").
The known sites (snps_dbsnp, snps_1000gp, known_indels, mills_indels) should be gzipped and tabix indexed.
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: ScatterFeatureRequirement
- class: SubworkflowFeatureRequirement
- class: MultipleInputFeatureRequirement
inputs:
- id: normal_inputs
doc: |-
An array of NORMAL FastqGz pairs. These are aligned separately and merged to create higher depth coverages from multiple sets of reads
type:
type: array
items:
type: array
items: File
- id: tumor_inputs
doc: |-
An array of TUMOR FastqGz pairs. These are aligned separately and merged to create higher depth coverages from multiple sets of reads
type:
type: array
items:
type: array
items: File
- id: normal_name
doc: |-
Sample name for the NORMAL sample from which to generate the readGroupHeaderLine for BwaMem
type: string
- id: tumor_name
doc: |-
Sample name for the TUMOR sample from which to generate the readGroupHeaderLine for BwaMem
type: string
- id: reference
doc: |2-
The reference genome from which to align the reads. This requires a number indexes (can be generated with the 'IndexFasta' pipeline This pipeline has been tested using the HG38 reference set.
This pipeline expects the assembly references to be as they appear in the GCP example. For example:
- HG38: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
- (".fai", ".amb", ".ann", ".bwt", ".pac", ".sa", "^.dict").
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: snps_dbsnp
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: snps_1000gp
doc: |-
From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
type: File
secondaryFiles:
- pattern: .tbi
- id: known_indels
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: mills_indels
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: gatk_intervals
doc: List of intervals over which to split the GATK variant calling
type:
type: array
items: File
- id: gridss_blacklist
doc: |-
BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist
type: File
- id: vardict_intervals
doc: List of intervals over which to split the VarDict variant calling
type:
type: array
items: File
- id: strelka_intervals
doc: An interval for which to restrict the analysis to.
type: File
secondaryFiles:
- pattern: .tbi
- id: gnomad
doc: |-
The genome Aggregation Database (gnomAD). This VCF must be compressed and tabix indexed. This is specific for your genome (eg: hg38 / br37) and can usually be found with your reference. For example for HG38, the Broad institute provide the following af-only-gnomad compressed and tabix indexed VCF: https://console.cloud.google.com/storage/browser/gatk-best-practices/somatic-hg38;tab=objects?prefix=af-only
type: File
secondaryFiles:
- pattern: .tbi
- id: panel_of_normals
doc: VCF file of sites observed in normal.
type:
- File
- 'null'
secondaryFiles:
- pattern: .tbi
- id: cutadapt_adapters
doc: |2-
Specifies a containment list for cutadapt, which contains a list of sequences to determine valid
overrepresented sequences from the FastQC report to trim with Cuatadapt. The file must contain sets
of named adapters in the form: ``name[tab]sequence``. Lines prefixed with a hash will be ignored.
type: File
- id: allele_freq_threshold
doc: "The threshold for VarDict's allele frequency, default: 0.05 or 5%"
type: float
default: 0.05
- id: combine_variants_type
doc: germline | somatic
type: string
default: somatic
- id: combine_variants_columns
doc: Columns to keep, seperated by space output vcf (unsorted)
type:
type: array
items: string
default:
- AD
- DP
- GT
outputs:
- id: out_normal_fastqc_reports
type:
type: array
items:
type: array
items: File
outputSource: normal/out_fastqc_reports
- id: out_tumor_fastqc_reports
type:
type: array
items:
type: array
items: File
outputSource: tumor/out_fastqc_reports
- id: out_normal_performance_summary
doc: A text file of performance summary of NORMAL bam
type: File
outputSource: normal/out_performance_summary
- id: out_tumor_performance_summary
doc: A text file of performance summary of TUMOR bam
type: File
outputSource: tumor/out_performance_summary
- id: out_normal_bam
type: File
secondaryFiles:
- pattern: .bai
outputSource: normal/out_bam
- id: out_tumor_bam
type: File
secondaryFiles:
- pattern: .bai
outputSource: tumor/out_bam
- id: out_gridss_assembly
doc: Assembly returned by GRIDSS
type: File
outputSource: vc_gridss/assembly
- id: out_variants_gridss
doc: Variants from the GRIDSS variant caller
type: File
outputSource: vc_gridss/out
- id: out_variants_gatk
doc: Merged variants from the GATK caller
type: File
outputSource: vc_gatk_sort_combined/out
- id: out_variants_split
doc: Unmerged variants from the GATK caller (by interval)
type:
type: array
items: File
outputSource: vc_gatk/out
- id: out_variants_vardict_split
doc: Unmerged variants from the VarDict caller (by interval)
type:
type: array
items: File
outputSource: vc_vardict/out
- id: out_variants_vardict
doc: Merged variants from the VarDict caller
type: File
outputSource: vc_vardict_sort_combined/out
- id: out_variants_strelka
doc: Variants from the Strelka variant caller
type: File
outputSource: vc_strelka/out
- id: out_variants
doc: Combined variants from GATK, VarDict and Strelka callers
type: File
outputSource: addbamstats/out
steps:
- id: tumor
in:
- id: reads
source: tumor_inputs
- id: sample_name
source: tumor_name
- id: reference
source: reference
- id: cutadapt_adapters
source: cutadapt_adapters
- id: gatk_intervals
source: gatk_intervals
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
run: tools/somatic_subpipeline.cwl
out:
- id: out_bam
- id: out_fastqc_reports
- id: out_performance_summary
- id: normal
in:
- id: reads
source: normal_inputs
- id: sample_name
source: normal_name
- id: reference
source: reference
- id: cutadapt_adapters
source: cutadapt_adapters
- id: gatk_intervals
source: gatk_intervals
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
run: tools/somatic_subpipeline.cwl
out:
- id: out_bam
- id: out_fastqc_reports
- id: out_performance_summary
- id: vc_gridss
label: Gridss
in:
- id: bams
source:
- normal/out_bam
- tumor/out_bam
- id: reference
source: reference
- id: blacklist
source: gridss_blacklist
run: tools/gridss_v2_6_2.cwl
out:
- id: out
- id: assembly
- id: bqsr_normal
label: GATK Base Recalibration on Bam
in:
- id: bam
source: normal/out_bam
- id: intervals
source: gatk_intervals
- id: reference
source: reference
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
scatter:
- intervals
run: tools/GATKBaseRecalBQSRWorkflow_4_1_3.cwl
out:
- id: out
- id: bqsr_tumor
label: GATK Base Recalibration on Bam
in:
- id: bam
source: tumor/out_bam
- id: intervals
source: gatk_intervals
- id: reference
source: reference
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
scatter:
- intervals
run: tools/GATKBaseRecalBQSRWorkflow_4_1_3.cwl
out:
- id: out
- id: vc_gatk
label: GATK4 Somatic Variant Caller
in:
- id: normal_bam
source: bqsr_normal/out
- id: tumor_bam
source: bqsr_tumor/out
- id: normal_name
source: normal_name
- id: intervals
source: gatk_intervals
- id: reference
source: reference
- id: gnomad
source: gnomad
- id: panel_of_normals
source: panel_of_normals
scatter:
- intervals
- normal_bam
- tumor_bam
scatterMethod: dotproduct
run: tools/GATK4_SomaticVariantCaller_4_1_3_0.cwl
out:
- id: variants
- id: out_bam
- id: out
- id: vc_gatk_merge
label: 'GATK4: Gather VCFs'
in:
- id: vcfs
source: vc_gatk/out
run: tools/Gatk4GatherVcfs_4_1_3_0.cwl
out:
- id: out
- id: vc_gatk_compress_for_sort
label: BGZip
in:
- id: file
source: vc_gatk_merge/out
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: vc_gatk_sort_combined
label: 'BCFTools: Sort'
in:
- id: vcf
source: vc_gatk_compress_for_sort/out
run: tools/bcftoolssort_v1_9.cwl
out:
- id: out
- id: vc_gatk_uncompress_for_combine
label: UncompressArchive
in:
- id: file
source: vc_gatk_sort_combined/out
run: tools/UncompressArchive_v1_0_0.cwl
out:
- id: out
- id: addbamstats
label: Annotate Bam Stats to Somatic Vcf Workflow
in:
- id: normal_id
source: normal_name
- id: tumor_id
source: tumor_name
- id: normal_bam
source: normal/out_bam
- id: tumor_bam
source: tumor/out_bam
- id: reference
source: reference
- id: vcf
source: vc_gatk_uncompress_for_combine/out
run: tools/AddBamStatsSomatic_v0_1_0.cwl
out:
- id: out
- id: generate_vardict_headerlines
label: GenerateVardictHeaderLines
in:
- id: reference
source: reference
run: tools/GenerateVardictHeaderLines_v0_1_0.cwl
out:
- id: out
- id: vc_vardict
label: Vardict Somatic Variant Caller
in:
- id: normal_bam
source: normal/out_bam
- id: tumor_bam
source: tumor/out_bam
- id: normal_name
source: normal_name
- id: tumor_name
source: tumor_name
- id: intervals
source: vardict_intervals
- id: allele_freq_threshold
source: allele_freq_threshold
- id: header_lines
source: generate_vardict_headerlines/out
- id: reference
source: reference
scatter:
- intervals
run: tools/vardictSomaticVariantCaller_v0_1_0.cwl
out:
- id: variants
- id: out
- id: vc_vardict_merge
label: 'GATK4: Gather VCFs'
in:
- id: vcfs
source: vc_vardict/out
run: tools/Gatk4GatherVcfs_4_1_3_0.cwl
out:
- id: out
- id: vc_vardict_compress_for_sort
label: BGZip
in:
- id: file
source: vc_vardict_merge/out
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: vc_vardict_sort_combined
label: 'BCFTools: Sort'
in:
- id: vcf
source: vc_vardict_compress_for_sort/out
run: tools/bcftoolssort_v1_9.cwl
out:
- id: out
- id: vc_vardict_uncompress_for_combine
label: UncompressArchive
in:
- id: file
source: vc_vardict_sort_combined/out
run: tools/UncompressArchive_v1_0_0.cwl
out:
- id: out
- id: vc_strelka
label: Strelka Somatic Variant Caller
in:
- id: normal_bam
source: normal/out_bam
- id: tumor_bam
source: tumor/out_bam
- id: reference
source: reference
- id: intervals
source: strelka_intervals
run: tools/strelkaSomaticVariantCaller_v0_1_1.cwl
out:
- id: sv
- id: variants
- id: out
- id: combine_variants
label: Combine Variants
in:
- id: vcfs
source:
- vc_gatk_uncompress_for_combine/out
- vc_strelka/out
- vc_vardict_uncompress_for_combine/out
- id: type
source: combine_variants_type
- id: columns
source: combine_variants_columns
- id: normal
source: normal_name
- id: tumor
source: tumor_name
run: tools/combinevariants_0_0_8.cwl
out:
- id: out
- id: combined_compress
label: BGZip
in:
- id: file
source: combine_variants/out
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: combined_sort
label: 'BCFTools: Sort'
in:
- id: vcf
source: combined_compress/out
run: tools/bcftoolssort_v1_9.cwl
out:
- id: out
- id: combined_uncompress
label: UncompressArchive
in:
- id: file
source: combined_sort/out
run: tools/UncompressArchive_v1_0_0.cwl
out:
- id: out
- id: combined_addbamstats
label: Annotate Bam Stats to Somatic Vcf Workflow
in:
- id: normal_id
source: normal_name
- id: tumor_id
source: tumor_name
- id: normal_bam
source: normal/out_bam
- id: tumor_bam
source: tumor/out_bam
- id: reference
source: reference
- id: vcf
source: combined_uncompress/out
run: tools/AddBamStatsSomatic_v0_1_0.cwl
out:
- id: out
id: WGSSomaticMultiCallers