Align and sort reads¶
BwaAligner · 1 contributor · 1 version
Align sorted bam with this subworkflow consisting of BWA Mem + SamTools + Gatk4SortSam
Quickstart¶
from janis_bioinformatics.tools.common.bwaaligner import BwaAligner wf = WorkflowBuilder("myworkflow") wf.step( "bwaaligner_step", BwaAligner( sample_name=None, reference=None, fastq=None, ) ) wf.output("out", source=bwaaligner_step.out)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for BwaAligner:
# user inputs
janis inputs BwaAligner > inputs.yaml
inputs.yaml
fastq:
- fastq_0.fastq.gz
- fastq_1.fastq.gz
reference: reference.fasta
sample_name: <value>
- Run BwaAligner with:
janis run [...run options] \
--inputs inputs.yaml \
BwaAligner
Information¶
URL: No URL to the documentation was provided
| ID: | BwaAligner |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | 1.0.0 |
| Authors: | Michael Franklin |
| Citations: | |
| Created: | 2018-12-24 |
| Updated: | None |
Outputs¶
| name | type | documentation |
|---|---|---|
| out | IndexedBam |
Workflow¶
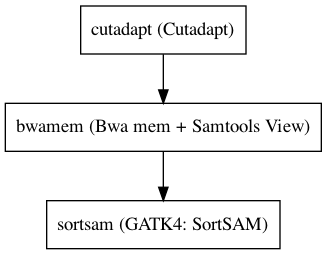
Embedded Tools¶
| Cutadapt | cutadapt/2.1 |
| Bwa mem + Samtools View | BwaMemSamtoolsView/0.7.17|1.9 |
| GATK4: SortSAM | Gatk4SortSam/4.1.2.0 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| sample_name | String | |
| reference | FastaWithIndexes | |
| fastq | FastqGzPair | |
| cutadapt_adapter | Optional<Array<String>> | |
| cutadapt_removeMiddle3Adapter | Optional<Array<String>> | |
| cutadapt_front | Optional<String> | (-g) Sequence of an adapter ligated to the 5’ end (paired data: of the first read). The adapter and any preceding bases are trimmed. Partial matches at the 5’ end are allowed. If a ‘^’ character is prepended (‘anchoring’), the adapter is only found if it is a prefix of the read. |
| cutadapt_removeMiddle5Adapter | Optional<String> | 5’ adapter to be removed from second read in a pair. |
| cutadapt_qualityCutoff | Optional<Integer> | (]3’CUTOFF, ]3’CUTOFF, -q) Trim low-quality bases from 5’ and/or 3’ ends of each read before adapter removal. Applied to both reads if data is paired. If one value is given, only the 3’ end is trimmed. If two comma-separated cutoffs are given, the 5’ end is trimmed with the first cutoff, the 3’ end with the second. |
| cutadapt_minimumLength | Optional<Integer> | (-m) Discard reads shorter than LEN. Default: 0 |
| bwamem_markShorterSplits | Optional<Boolean> | Mark shorter split hits as secondary (for Picard compatibility). |
| sortsam_sortOrder | Optional<String> | The –SORT_ORDER argument is an enumerated type (SortOrder), which can have one of the following values: [unsorted, queryname, coordinate, duplicate, unknown] |
| sortsam_createIndex | Optional<Boolean> | Whether to create a BAM index when writing a coordinate-sorted BAM file. |
| sortsam_validationStringency | Optional<String> | Validation stringency for all SAM files read by this program. Setting stringency to SILENT can improve performance when processing a BAM file in which variable-length data (read, qualities, tags) do not otherwise need to be decoded.The –VALIDATION_STRINGENCY argument is an enumerated type (ValidationStringency), which can have one of the following values: [STRICT, LENIENT, SILENT] |
| sortsam_maxRecordsInRam | Optional<Integer> | When writing SAM files that need to be sorted, this will specify the number of records stored in RAM before spilling to disk. Increasing this number reduces the number of file handles needed to sort a SAM file, and increases the amount of RAM needed. |
| sortsam_tmpDir | Optional<String> | Undocumented option |
Workflow Description Language¶
version development
import "tools/cutadapt_2_1.wdl" as C
import "tools/BwaMemSamtoolsView_0_7_17_1_9.wdl" as B
import "tools/Gatk4SortSam_4_1_2_0.wdl" as G
workflow BwaAligner {
input {
String sample_name
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
Array[File] fastq
Array[String]? cutadapt_adapter
Array[String]? cutadapt_removeMiddle3Adapter
String? cutadapt_front
String? cutadapt_removeMiddle5Adapter
Int? cutadapt_qualityCutoff = 15
Int? cutadapt_minimumLength = 50
Boolean? bwamem_markShorterSplits = true
String? sortsam_sortOrder = "coordinate"
Boolean? sortsam_createIndex = true
String? sortsam_validationStringency = "SILENT"
Int? sortsam_maxRecordsInRam = 5000000
String? sortsam_tmpDir = "."
}
call C.cutadapt as cutadapt {
input:
outputPrefix=sample_name,
fastq=fastq,
adapter=cutadapt_adapter,
front=cutadapt_front,
qualityCutoff=select_first([cutadapt_qualityCutoff, 15]),
minimumLength=select_first([cutadapt_minimumLength, 50]),
removeMiddle3Adapter=cutadapt_removeMiddle3Adapter,
removeMiddle5Adapter=cutadapt_removeMiddle5Adapter
}
call B.BwaMemSamtoolsView as bwamem {
input:
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
reads=cutadapt.out,
sampleName=sample_name,
markShorterSplits=select_first([bwamem_markShorterSplits, true])
}
call G.Gatk4SortSam as sortsam {
input:
bam=bwamem.out,
sortOrder=select_first([sortsam_sortOrder, "coordinate"]),
createIndex=select_first([sortsam_createIndex, true]),
maxRecordsInRam=select_first([sortsam_maxRecordsInRam, 5000000]),
tmpDir=select_first([sortsam_tmpDir, "."]),
validationStringency=select_first([sortsam_validationStringency, "SILENT"])
}
output {
File out = sortsam.out
File out_bai = sortsam.out_bai
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Align and sort reads
doc: |-
Align sorted bam with this subworkflow consisting of BWA Mem + SamTools + Gatk4SortSam
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
inputs:
- id: sample_name
type: string
- id: reference
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: fastq
type:
type: array
items: File
- id: cutadapt_adapter
type:
- type: array
items: string
- 'null'
- id: cutadapt_removeMiddle3Adapter
type:
- type: array
items: string
- 'null'
- id: cutadapt_front
doc: |-
(-g) Sequence of an adapter ligated to the 5' end (paired data: of the first read). The adapter and any preceding bases are trimmed. Partial matches at the 5' end are allowed. If a '^' character is prepended ('anchoring'), the adapter is only found if it is a prefix of the read.
type:
- string
- 'null'
- id: cutadapt_removeMiddle5Adapter
doc: 5' adapter to be removed from second read in a pair.
type:
- string
- 'null'
- id: cutadapt_qualityCutoff
doc: |-
(]3'CUTOFF, ]3'CUTOFF, -q) Trim low-quality bases from 5' and/or 3' ends of each read before adapter removal. Applied to both reads if data is paired. If one value is given, only the 3' end is trimmed. If two comma-separated cutoffs are given, the 5' end is trimmed with the first cutoff, the 3' end with the second.
type: int
default: 15
- id: cutadapt_minimumLength
doc: '(-m) Discard reads shorter than LEN. Default: 0'
type: int
default: 50
- id: bwamem_markShorterSplits
doc: Mark shorter split hits as secondary (for Picard compatibility).
type: boolean
default: true
- id: sortsam_sortOrder
doc: |-
The --SORT_ORDER argument is an enumerated type (SortOrder), which can have one of the following values: [unsorted, queryname, coordinate, duplicate, unknown]
type: string
default: coordinate
- id: sortsam_createIndex
doc: Whether to create a BAM index when writing a coordinate-sorted BAM file.
type: boolean
default: true
- id: sortsam_validationStringency
doc: |-
Validation stringency for all SAM files read by this program. Setting stringency to SILENT can improve performance when processing a BAM file in which variable-length data (read, qualities, tags) do not otherwise need to be decoded.The --VALIDATION_STRINGENCY argument is an enumerated type (ValidationStringency), which can have one of the following values: [STRICT, LENIENT, SILENT]
type: string
default: SILENT
- id: sortsam_maxRecordsInRam
doc: |-
When writing SAM files that need to be sorted, this will specify the number of records stored in RAM before spilling to disk. Increasing this number reduces the number of file handles needed to sort a SAM file, and increases the amount of RAM needed.
type: int
default: 5000000
- id: sortsam_tmpDir
doc: Undocumented option
type: string
default: .
outputs:
- id: out
type: File
secondaryFiles:
- pattern: .bai
outputSource: sortsam/out
steps:
- id: cutadapt
label: Cutadapt
in:
- id: outputPrefix
source: sample_name
- id: fastq
source: fastq
- id: adapter
source: cutadapt_adapter
- id: front
source: cutadapt_front
- id: qualityCutoff
source: cutadapt_qualityCutoff
- id: minimumLength
source: cutadapt_minimumLength
- id: removeMiddle3Adapter
source: cutadapt_removeMiddle3Adapter
- id: removeMiddle5Adapter
source: cutadapt_removeMiddle5Adapter
run: tools/cutadapt_2_1.cwl
out:
- id: out
- id: bwamem
label: Bwa mem + Samtools View
in:
- id: reference
source: reference
- id: reads
source: cutadapt/out
- id: sampleName
source: sample_name
- id: markShorterSplits
source: bwamem_markShorterSplits
run: tools/BwaMemSamtoolsView_0_7_17_1_9.cwl
out:
- id: out
- id: sortsam
label: 'GATK4: SortSAM'
in:
- id: bam
source: bwamem/out
- id: sortOrder
source: sortsam_sortOrder
- id: createIndex
source: sortsam_createIndex
- id: maxRecordsInRam
source: sortsam_maxRecordsInRam
- id: tmpDir
source: sortsam_tmpDir
- id: validationStringency
source: sortsam_validationStringency
run: tools/Gatk4SortSam_4_1_2_0.cwl
out:
- id: out
id: BwaAligner