Oncopipe: sample preparation¶
OncopipeSamplePreparation · 1 contributor · 1 version
No documentation was provided: contribute one
Quickstart¶
from janis_bioinformatics.tools.oshlack.oncopipe.oncopipe import OncopipeSamplePreparation wf = WorkflowBuilder("myworkflow") wf.step( "oncopipesamplepreparation_step", OncopipeSamplePreparation( name=None, reads=None, genome_dir=None, reference=None, gtf=None, blacklist=None, ) ) wf.output("out_arriba_bam", source=oncopipesamplepreparation_step.out_arriba_bam) wf.output("out_arriba_fusion", source=oncopipesamplepreparation_step.out_arriba_fusion) wf.output("out_arriba_fusion_discarded", source=oncopipesamplepreparation_step.out_arriba_fusion_discarded) wf.output("out_gene_counts", source=oncopipesamplepreparation_step.out_gene_counts) wf.output("out_predictions", source=oncopipesamplepreparation_step.out_predictions) wf.output("out_probabilities", source=oncopipesamplepreparation_step.out_probabilities) wf.output("out_distributions", source=oncopipesamplepreparation_step.out_distributions) wf.output("out_waterfalls", source=oncopipesamplepreparation_step.out_waterfalls)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for OncopipeSamplePreparation:
# user inputs
janis inputs OncopipeSamplePreparation > inputs.yaml
inputs.yaml
blacklist: blacklist
genome_dir: null
gtf: gtf
name: <value>
reads:
- reads_0.fastq.gz
- reads_1.fastq.gz
reference: reference.fasta
- Run OncopipeSamplePreparation with:
janis run [...run options] \
--inputs inputs.yaml \
OncopipeSamplePreparation
Information¶
URL: No URL to the documentation was provided
| ID: | OncopipeSamplePreparation |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | v0.1.0 |
| Authors: | Michael Franklin |
| Citations: | |
| Created: | 2020-09-24 |
| Updated: | 2020-10-07 |
Outputs¶
| name | type | documentation |
|---|---|---|
| out_arriba_bam | IndexedBam | |
| out_arriba_fusion | tsv | |
| out_arriba_fusion_discarded | tsv | |
| out_gene_counts | TextFile | |
| out_predictions | csv | |
| out_probabilities | csv | |
| out_distributions | File | |
| out_waterfalls | File |
Workflow¶
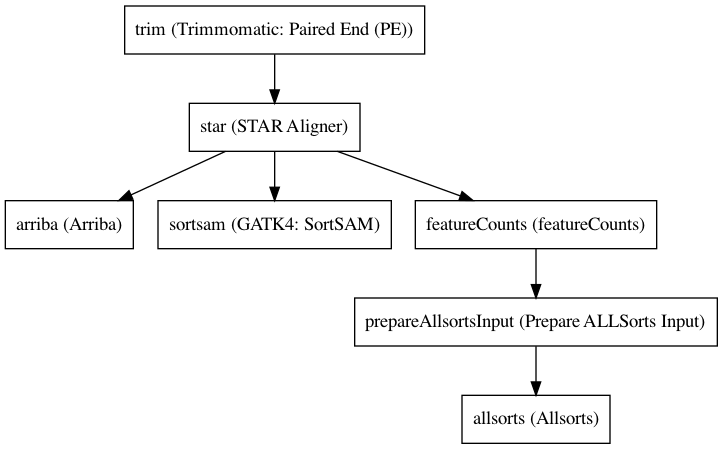
Embedded Tools¶
| Trimmomatic: Paired End (PE) | trimmomaticPairedEnd/0.35 |
| STAR Aligner | star_alignReads/v2.7.1a |
| Arriba | Arriba/1.2.0 |
| GATK4: SortSAM | Gatk4SortSam/4.1.4.0 |
| featureCounts | featureCounts/2.0.1 |
| Prepare ALLSorts Input | prepareALLSortsInput/v0.1.0 |
| Allsorts | Allsorts/v0.1.0 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| name | String | Sample ID |
| reads | FastqGzPair | |
| genome_dir | Directory | |
| reference | Fasta | |
| gtf | File | |
| blacklist | File | |
| contigs | Optional<Array<String>> | |
| trim_phred33 | Optional<Boolean> | Use phred + 33 quality score. If no quality encoding is specified, it will be determined automatically |
| trim_steps | Optional<Array<String>> | ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read. SLIDINGWINDOW: Performs a sliding window trimming approach. It starts scanning at the 5” end and clips the read once the average quality within the window falls below a threshold. MAXINFO: An adaptive quality trimmer which balances read length and error rate to maximise the value of each read LEADING: Cut bases off the start of a read, if below a threshold quality TRAILING: Cut bases off the end of a read, if below a threshold quality CROP: Cut the read to a specified length by removing bases from the end HEADCROP: Cut the specified number of bases from the start of the read MINLEN: Drop the read if it is below a specified length AVGQUAL: Drop the read if the average quality is below the specified level TOPHRED33: Convert quality scores to Phred-33 TOPHRED64: Convert quality scores to Phred-64 |
| star_limitOutSJcollapsed | Optional<Integer> | (default: 1000000) max number of collapsed junctions |
| star_readFilesCommand | Optional<String> | (default: -) command line to execute for each of the input file. This command should generate FASTA or FASTQ text and send it to stdout zcat - to uncompress .gz files, bzcat - to uncompress .bz2 files, etc. |
| star_outSAMtype | Optional<Array<String>> | (default: SAM) … quasi-random order used before 2.5.0 Random … random order of alignments for each multi-mapper. Read mates (pairs) are always adjacent, all alignment for each read stay together. This option will become default in the future releases. … standard unsorted SortedByCoordinate … sorted by coordinate. This option will allocate extra memory for sorting which can be specified by –limitBAMsortRAM. |
| star_outSAMunmapped | Optional<String> | (default: None) output of unmapped reads in the SAM format 1st word: None … no output Within … output unmapped reads within the main SAM file (i.e. Aligned.out.sam) 2nd word: KeepPairs … record unmapped mate for each alignment, and, in case of unsorted output, keep it adjacent to its mapped mate. Only affects multi-mapping reads. |
| star_outBAMcompression | Optional<Integer> | (default: 1) -1 to 10 BAM compression level, -1=default compression (6?), 0=no compression, 10=maximum compression |
| star_outFilterMultimapNmax | Optional<Integer> | (default: 10) maximum number of loci the read is allowed to map to. Alignments (all of them) will be output only if the read maps to no more loci than this value. Otherwise no alignments will be output, and the read will be counted as “mapped to too many loci” in the Log.final.out . |
| star_outFilterMismatchNmax | Optional<Integer> | (default: 10) alignment will be output only if it has no more mismatches than this value. |
| star_chimSegmentMin | Optional<Integer> | (default: 0) minimum length of chimeric segment length, if ==0, no chimeric output |
| star_chimOutType | Optional<Array<String>> | (default: Junctions) type of chimeric output Junctions … Chimeric.out.junction SeparateSAMold … output old SAM into separate Chimeric.out.sam file WithinBAM … output into main aligned BAM files (Aligned.*.bam) WithinBAM HardClip … (default) hard-clipping in the CIGAR for supplemental chimeric alignments (defaultif no 2nd word is present) WithinBAM SoftClip … soft-clipping in the CIGAR for supplemental chimeric alignments |
| star_chimJunctionOverhangMin | Optional<Integer> | (default: 20) minimum overhang for a chimeric junction |
| star_chimScoreMin | Optional<Integer> | (default: 0) minimum total (summed) score of the chimeric segments |
| star_chimScoreDropMax | Optional<Integer> | (default: 20) max drop (difference) of chimeric score (the sum of scores of all chimeric segments) from the read length |
| star_chimScoreJunctionNonGTAG | Optional<Integer> | (default: -1) penalty for a non-GT/AG chimeric junction |
| star_chimScoreSeparation | Optional<Integer> | (default: 10) minimum difference (separation) between the best chimeric score and the next one |
| star_alignSJstitchMismatchNmax | Optional<Array<Integer>> | (default: 0 -1 0 0) maximum number of mismatches for stitching of the splice junctions (-1: no limit). (1) non-canonical motifs, (2) GT/AG and CT/AC motif, (3) GC/AG and CT/GC motif, (4) AT/AC and GT/AT motif. |
| star_chimSegmentReadGapMax | Optional<Integer> | (default: 0) maximum gap in the read sequence between chimeric segments |
| arriba_fusion_transcript | Optional<Boolean> | When set, the column ‘fusion_transcript’ is populated with the sequence of the fused genes as assembled from the supporting reads. Specify the flag twice to also print the fusion transcripts to the file containing discarded fusions (-O). Default: off |
| arriba_peptide_sequence | Optional<Boolean> | When set, the column ‘peptide_sequence’ is populated with the sequence of the fused proteins as assembled from the supporting reads. Specify the flag twice to also print the peptide sequence to the file containing discarded fusions (-O). Default: off |
| sortsam_sortOrder | Optional<String> | The –SORT_ORDER argument is an enumerated type (SortOrder), which can have one of the following values: [unsorted, queryname, coordinate, duplicate, unknown] |
| sortsam_createIndex | Optional<Boolean> | Whether to create a BAM index when writing a coordinate-sorted BAM file. |
| featureCounts_attributeType | Optional<String> | Specify attribute type in GTF annotation. ‘gene_id’ by default. Meta-features used for read counting will be extracted from annotation using the provided value. |
| prepareAllsortsInput_fusion_caller | Optional<String> |
Workflow Description Language¶
version development
import "tools/trimmomaticPairedEnd_0_35.wdl" as T
import "tools/star_alignReads_v2_7_1a.wdl" as S
import "tools/Arriba_1_2_0.wdl" as A
import "tools/Gatk4SortSam_4_1_4_0.wdl" as G
import "tools/featureCounts_2_0_1.wdl" as F
import "tools/prepareALLSortsInput_v0_1_0.wdl" as P
import "tools/Allsorts_v0_1_0.wdl" as A2
workflow OncopipeSamplePreparation {
input {
String name
Array[File] reads
Directory genome_dir
File reference
File gtf
File blacklist
Array[String]? contigs
Boolean? trim_phred33 = true
Array[String]? trim_steps = ["ILLUMINACLIP:/usr/local/share/trimmomatic-0.35-6/adapters/TruSeq2-PE.fa:2:30:10", "LEADING:15", "TRAILING:15", "SLIDINGWINDOW:4:15", "MINLEN:35"]
Int? star_limitOutSJcollapsed = 3000000
String? star_readFilesCommand = "zcat"
Array[String]? star_outSAMtype = ["BAM", "Unsorted"]
String? star_outSAMunmapped = "Within"
Int? star_outBAMcompression = 0
Int? star_outFilterMultimapNmax = 1
Int? star_outFilterMismatchNmax = 3
Int? star_chimSegmentMin = 10
Array[String]? star_chimOutType = ["WithinBAM", "SoftClip"]
Int? star_chimJunctionOverhangMin = 10
Int? star_chimScoreMin = 1
Int? star_chimScoreDropMax = 30
Int? star_chimScoreJunctionNonGTAG = 0
Int? star_chimScoreSeparation = 1
Array[Int]? star_alignSJstitchMismatchNmax = [5, -1, 5, 5]
Int? star_chimSegmentReadGapMax = 3
Boolean? arriba_fusion_transcript = true
Boolean? arriba_peptide_sequence = true
String? sortsam_sortOrder = "coordinate"
Boolean? sortsam_createIndex = true
String? featureCounts_attributeType = "gene_name"
String? prepareAllsortsInput_fusion_caller = "featureCounts"
}
call T.trimmomaticPairedEnd as trim {
input:
steps=select_first([trim_steps, ["ILLUMINACLIP:/usr/local/share/trimmomatic-0.35-6/adapters/TruSeq2-PE.fa:2:30:10", "LEADING:15", "TRAILING:15", "SLIDINGWINDOW:4:15", "MINLEN:35"]]),
sampleName=name,
phred33=select_first([trim_phred33, true]),
inp=reads
}
call S.star_alignReads as star {
input:
genomeDir=genome_dir,
readFilesIn=trim.pairedOut,
readFilesCommand=select_first([star_readFilesCommand, "zcat"]),
limitOutSJcollapsed=select_first([star_limitOutSJcollapsed, 3000000]),
outSAMtype=select_first([star_outSAMtype, ["BAM", "Unsorted"]]),
outSAMunmapped=select_first([star_outSAMunmapped, "Within"]),
outBAMcompression=select_first([star_outBAMcompression, 0]),
outFilterMultimapNmax=select_first([star_outFilterMultimapNmax, 1]),
outFilterMismatchNmax=select_first([star_outFilterMismatchNmax, 3]),
alignSJstitchMismatchNmax=select_first([star_alignSJstitchMismatchNmax, [5, -1, 5, 5]]),
chimOutType=select_first([star_chimOutType, ["WithinBAM", "SoftClip"]]),
chimSegmentMin=select_first([star_chimSegmentMin, 10]),
chimScoreMin=select_first([star_chimScoreMin, 1]),
chimScoreDropMax=select_first([star_chimScoreDropMax, 30]),
chimScoreSeparation=select_first([star_chimScoreSeparation, 1]),
chimScoreJunctionNonGTAG=select_first([star_chimScoreJunctionNonGTAG, 0]),
chimJunctionOverhangMin=select_first([star_chimJunctionOverhangMin, 10]),
chimSegmentReadGapMax=select_first([star_chimSegmentReadGapMax, 3])
}
call A.Arriba as arriba {
input:
aligned_inp=select_first([star.out_unsorted_bam]),
gtf_file=gtf,
reference=reference,
blacklist=blacklist,
contigs=contigs,
fusion_transcript=select_first([arriba_fusion_transcript, true]),
peptide_sequence=select_first([arriba_peptide_sequence, true])
}
call G.Gatk4SortSam as sortsam {
input:
bam=select_first([star.out_unsorted_bam]),
sortOrder=select_first([sortsam_sortOrder, "coordinate"]),
createIndex=select_first([sortsam_createIndex, true])
}
call F.featureCounts as featureCounts {
input:
attributeType=select_first([featureCounts_attributeType, "gene_name"]),
bam=[select_first([star.out_unsorted_bam])],
annotationFile=gtf
}
call P.prepareALLSortsInput as prepareAllsortsInput {
input:
inps=[featureCounts.out],
labels=[name],
fusion_caller=select_first([prepareAllsortsInput_fusion_caller, "featureCounts"])
}
call A2.Allsorts as allsorts {
input:
samples=prepareAllsortsInput.out
}
output {
File out_arriba_bam = sortsam.out
File out_arriba_bam_bai = sortsam.out_bai
File out_arriba_fusion = arriba.out
File out_arriba_fusion_discarded = arriba.out_discarded
File out_gene_counts = featureCounts.out
File out_predictions = allsorts.out_predictions
File out_probabilities = allsorts.out_probabilities
File out_distributions = allsorts.out_distributions
File out_waterfalls = allsorts.out_waterfalls
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: 'Oncopipe: sample preparation'
doc: ''
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: MultipleInputFeatureRequirement
inputs:
- id: name
doc: Sample ID
type: string
- id: reads
type:
type: array
items: File
- id: genome_dir
type: Directory
- id: reference
type: File
- id: gtf
type: File
- id: blacklist
type: File
- id: contigs
type:
- type: array
items: string
- 'null'
- id: trim_phred33
doc: |-
Use phred + 33 quality score. If no quality encoding is specified, it will be determined automatically
type: boolean
default: true
- id: trim_steps
doc: |
ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read.
SLIDINGWINDOW: Performs a sliding window trimming approach. It starts
scanning at the 5" end and clips the read once the average quality within the window
falls below a threshold.
MAXINFO: An adaptive quality trimmer which balances read length and error rate to
maximise the value of each read
LEADING: Cut bases off the start of a read, if below a threshold quality
TRAILING: Cut bases off the end of a read, if below a threshold quality
CROP: Cut the read to a specified length by removing bases from the end
HEADCROP: Cut the specified number of bases from the start of the read
MINLEN: Drop the read if it is below a specified length
AVGQUAL: Drop the read if the average quality is below the specified level
TOPHRED33: Convert quality scores to Phred-33
TOPHRED64: Convert quality scores to Phred-64
type:
type: array
items: string
default:
- ILLUMINACLIP:/usr/local/share/trimmomatic-0.35-6/adapters/TruSeq2-PE.fa:2:30:10
- LEADING:15
- TRAILING:15
- SLIDINGWINDOW:4:15
- MINLEN:35
- id: star_limitOutSJcollapsed
doc: '(default: 1000000) max number of collapsed junctions'
type: int
default: 3000000
- id: star_readFilesCommand
doc: |-
(default: -) command line to execute for each of the input file. This command should generate FASTA or FASTQ text and send it to stdout zcat - to uncompress .gz files, bzcat - to uncompress .bz2 files, etc.
type: string
default: zcat
- id: star_outSAMtype
doc: |-
(default: SAM) ... quasi-random order used before 2.5.0 Random ... random order of alignments for each multi-mapper. Read mates (pairs) are always adjacent, all alignment for each read stay together. This option will become default in the future releases. ... standard unsorted SortedByCoordinate ... sorted by coordinate. This option will allocate extra memory for sorting which can be specified by --limitBAMsortRAM.
type:
type: array
items: string
default:
- BAM
- Unsorted
- id: star_outSAMunmapped
doc: |-
(default: None) output of unmapped reads in the SAM format 1st word: None ... no output Within ... output unmapped reads within the main SAM file (i.e. Aligned.out.sam) 2nd word: KeepPairs ... record unmapped mate for each alignment, and, in case of unsorted output, keep it adjacent to its mapped mate. Only affects multi-mapping reads.
type: string
default: Within
- id: star_outBAMcompression
doc: |-
(default: 1) -1 to 10 BAM compression level, -1=default compression (6?), 0=no compression, 10=maximum compression
type: int
default: 0
- id: star_outFilterMultimapNmax
doc: |-
(default: 10) maximum number of loci the read is allowed to map to. Alignments (all of them) will be output only if the read maps to no more loci than this value. Otherwise no alignments will be output, and the read will be counted as "mapped to too many loci" in the Log.final.out .
type: int
default: 1
- id: star_outFilterMismatchNmax
doc: |-
(default: 10) alignment will be output only if it has no more mismatches than this value.
type: int
default: 3
- id: star_chimSegmentMin
doc: |-
(default: 0) minimum length of chimeric segment length, if ==0, no chimeric output
type: int
default: 10
- id: star_chimOutType
doc: |-
(default: Junctions) type of chimeric output Junctions ... Chimeric.out.junction SeparateSAMold ... output old SAM into separate Chimeric.out.sam file WithinBAM ... output into main aligned BAM files (Aligned.*.bam) WithinBAM HardClip ... (default) hard-clipping in the CIGAR for supplemental chimeric alignments (defaultif no 2nd word is present) WithinBAM SoftClip ... soft-clipping in the CIGAR for supplemental chimeric alignments
type:
type: array
items: string
default:
- WithinBAM
- SoftClip
- id: star_chimJunctionOverhangMin
doc: '(default: 20) minimum overhang for a chimeric junction'
type: int
default: 10
- id: star_chimScoreMin
doc: '(default: 0) minimum total (summed) score of the chimeric segments'
type: int
default: 1
- id: star_chimScoreDropMax
doc: |-
(default: 20) max drop (difference) of chimeric score (the sum of scores of all chimeric segments) from the read length
type: int
default: 30
- id: star_chimScoreJunctionNonGTAG
doc: '(default: -1) penalty for a non-GT/AG chimeric junction'
type: int
default: 0
- id: star_chimScoreSeparation
doc: |-
(default: 10) minimum difference (separation) between the best chimeric score and the next one
type: int
default: 1
- id: star_alignSJstitchMismatchNmax
doc: |-
(default: 0 -1 0 0) maximum number of mismatches for stitching of the splice junctions (-1: no limit). (1) non-canonical motifs, (2) GT/AG and CT/AC motif, (3) GC/AG and CT/GC motif, (4) AT/AC and GT/AT motif.
type:
type: array
items: int
default:
- 5
- -1
- 5
- 5
- id: star_chimSegmentReadGapMax
doc: '(default: 0) maximum gap in the read sequence between chimeric segments'
type: int
default: 3
- id: arriba_fusion_transcript
doc: |-
When set, the column 'fusion_transcript' is populated with the sequence of the fused genes as assembled from the supporting reads. Specify the flag twice to also print the fusion transcripts to the file containing discarded fusions (-O). Default: off
type: boolean
default: true
- id: arriba_peptide_sequence
doc: |-
When set, the column 'peptide_sequence' is populated with the sequence of the fused proteins as assembled from the supporting reads. Specify the flag twice to also print the peptide sequence to the file containing discarded fusions (-O). Default: off
type: boolean
default: true
- id: sortsam_sortOrder
doc: |-
The --SORT_ORDER argument is an enumerated type (SortOrder), which can have one of the following values: [unsorted, queryname, coordinate, duplicate, unknown]
type: string
default: coordinate
- id: sortsam_createIndex
doc: Whether to create a BAM index when writing a coordinate-sorted BAM file.
type: boolean
default: true
- id: featureCounts_attributeType
doc: |-
Specify attribute type in GTF annotation. 'gene_id' by default. Meta-features used for read counting will be extracted from annotation using the provided value.
type: string
default: gene_name
- id: prepareAllsortsInput_fusion_caller
type: string
default: featureCounts
outputs:
- id: out_arriba_bam
type: File
secondaryFiles:
- pattern: .bai
outputSource: sortsam/out
- id: out_arriba_fusion
type: File
outputSource: arriba/out
- id: out_arriba_fusion_discarded
type: File
outputSource: arriba/out_discarded
- id: out_gene_counts
type: File
outputSource: featureCounts/out
- id: out_predictions
type: File
outputSource: allsorts/out_predictions
- id: out_probabilities
type: File
outputSource: allsorts/out_probabilities
- id: out_distributions
type: File
outputSource: allsorts/out_distributions
- id: out_waterfalls
type: File
outputSource: allsorts/out_waterfalls
steps:
- id: trim
label: 'Trimmomatic: Paired End (PE)'
doc: Trim reads using Trimmomatic
in:
- id: steps
source: trim_steps
- id: sampleName
source: name
- id: phred33
source: trim_phred33
- id: inp
source: reads
run: tools/trimmomaticPairedEnd_0_35.cwl
out:
- id: pairedOut
- id: unpairedOut
- id: star
label: STAR Aligner
in:
- id: genomeDir
source: genome_dir
- id: readFilesIn
source: trim/pairedOut
- id: readFilesCommand
source: star_readFilesCommand
- id: limitOutSJcollapsed
source: star_limitOutSJcollapsed
- id: outSAMtype
source: star_outSAMtype
- id: outSAMunmapped
source: star_outSAMunmapped
- id: outBAMcompression
source: star_outBAMcompression
- id: outFilterMultimapNmax
source: star_outFilterMultimapNmax
- id: outFilterMismatchNmax
source: star_outFilterMismatchNmax
- id: alignSJstitchMismatchNmax
source: star_alignSJstitchMismatchNmax
- id: chimOutType
source: star_chimOutType
- id: chimSegmentMin
source: star_chimSegmentMin
- id: chimScoreMin
source: star_chimScoreMin
- id: chimScoreDropMax
source: star_chimScoreDropMax
- id: chimScoreSeparation
source: star_chimScoreSeparation
- id: chimScoreJunctionNonGTAG
source: star_chimScoreJunctionNonGTAG
- id: chimJunctionOverhangMin
source: star_chimJunctionOverhangMin
- id: chimSegmentReadGapMax
source: star_chimSegmentReadGapMax
run: tools/star_alignReads_v2_7_1a.cwl
out:
- id: out_unsorted_bam
- id: out_sorted_bam
- id: SJ_out_tab
- id: Log_out
- id: Log_progress_out
- id: Log_final_out
- id: arriba
label: Arriba
in:
- id: _arriba_aligned_inp_staroutunsortedbam
source: star/out_unsorted_bam
- id: aligned_inp
valueFrom: $(inputs._arriba_aligned_inp_staroutunsortedbam)
- id: gtf_file
source: gtf
- id: reference
source: reference
- id: blacklist
source: blacklist
- id: contigs
source: contigs
- id: fusion_transcript
source: arriba_fusion_transcript
- id: peptide_sequence
source: arriba_peptide_sequence
run: tools/Arriba_1_2_0.cwl
out:
- id: out
- id: out_discarded
- id: sortsam
label: 'GATK4: SortSAM'
in:
- id: _sortsam_bam_staroutunsortedbam
source: star/out_unsorted_bam
- id: bam
valueFrom: $(inputs._sortsam_bam_staroutunsortedbam)
- id: sortOrder
source: sortsam_sortOrder
- id: createIndex
source: sortsam_createIndex
run: tools/Gatk4SortSam_4_1_4_0.cwl
out:
- id: out
- id: featureCounts
label: featureCounts
in:
- id: attributeType
source: featureCounts_attributeType
- id: _featureCounts_bam_staroutunsortedbam
source: star/out_unsorted_bam
- id: bam
valueFrom: $([inputs._featureCounts_bam_staroutunsortedbam])
- id: annotationFile
source: gtf
run: tools/featureCounts_2_0_1.cwl
out:
- id: out
- id: prepareAllsortsInput
label: Prepare ALLSorts Input
in:
- id: inps
source:
- featureCounts/out
linkMerge: merge_nested
- id: labels
source:
- name
linkMerge: merge_nested
- id: fusion_caller
source: prepareAllsortsInput_fusion_caller
run: tools/prepareALLSortsInput_v0_1_0.cwl
out:
- id: out
- id: allsorts
label: Allsorts
in:
- id: samples
source: prepareAllsortsInput/out
run: tools/Allsorts_v0_1_0.cwl
out:
- id: out_predictions
- id: out_probabilities
- id: out_distributions
- id: out_waterfalls
id: OncopipeSamplePreparation