WGS Germline (GATK) [VARIANTS only]¶
WGSGermlineGATKVariantsOnly · A variant-calling WGS pipeline using only the GATK Haplotype variant caller · 3 contributors · 1 version
This is a genomics pipeline to ONLY call variants using GATK and GRIDSS from an indexed bam. The final variants are outputted in the VCF format.
This workflow is a reference pipeline using the Janis Python framework (pipelines assistant).
- Call variants using GRIDSS and GATK4;
- Outputs the final variants in the VCF format.
Resources
This pipeline has been tested using the HG38 reference set, available on Google Cloud Storage through:
This pipeline expects the assembly references to be as they appear in that storage (“.fai”, “.amb”, “.ann”, “.bwt”, “.pac”, “.sa”, “^.dict”). The known sites (snps_dbsnp, snps_1000gp, known_indels, mills_indels) should be gzipped and tabix indexed.
Quickstart¶
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
| Name | Type | Source | Description |
|---|---|---|---|
| reference | FastaWithIndexes |
|
The reference genome from which to align the reads. This requires a number indexes (can be generated with the ‘IndexFasta’ pipeline This pipeline has been tested using the HG38 reference set.
|
| snps_dbsnp | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| snps_1000gp | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/ |
| known_indels | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| mills_indels | Gzipped<VCF> |
|
From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| gridss_blacklist | bed | BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist | |
| gatk_intervals | Array<bed> | None | List of intervals over which to split the GATK variant calling |
- Generate user and static input files for WGSGermlineGATKVariantsOnly:
# user inputs
janis inputs --user WGSGermlineGATKVariantsOnly > inputs.yaml
# static inputs
janis inputs --static WGSGermlineGATKVariantsOnly > static.yaml
inputs.yaml
bam: NA12878.bam
sample_name: NA12878
static.yaml
gatk_intervals: BRCA1.bed
gridss_blacklist: gridss_blacklist.bed
known_indels: Homo_sapiens_assembly38.known_indels.vcf.gz
mills_indels: Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
reference: Homo_sapiens_assembly38.fasta
snps_1000gp: 1000G_phase1.snps.high_confidence.hg38.vcf.gz
snps_dbsnp: Homo_sapiens_assembly38.dbsnp138.vcf.gz
- Run WGSGermlineGATKVariantsOnly with:
janis run [...run options] \
--inputs inputs.yaml \
--inputs static.yaml \
WGSGermlineGATKVariantsOnly
Outputs¶
| name | type | documentation |
|---|---|---|
| out_performance_summary | csv | A text file of performance summary of bam |
| out_gridss_assembly | BAM | Assembly returned by GRIDSS |
| out_variants_gridss | VCF | Variants from the GRIDSS variant caller |
| out_variants | Gzipped<VCF> | Merged variants from the GATK caller |
| out_variants_split | Array<VCF> | Unmerged variants from the GATK caller (by interval) |
Workflow¶
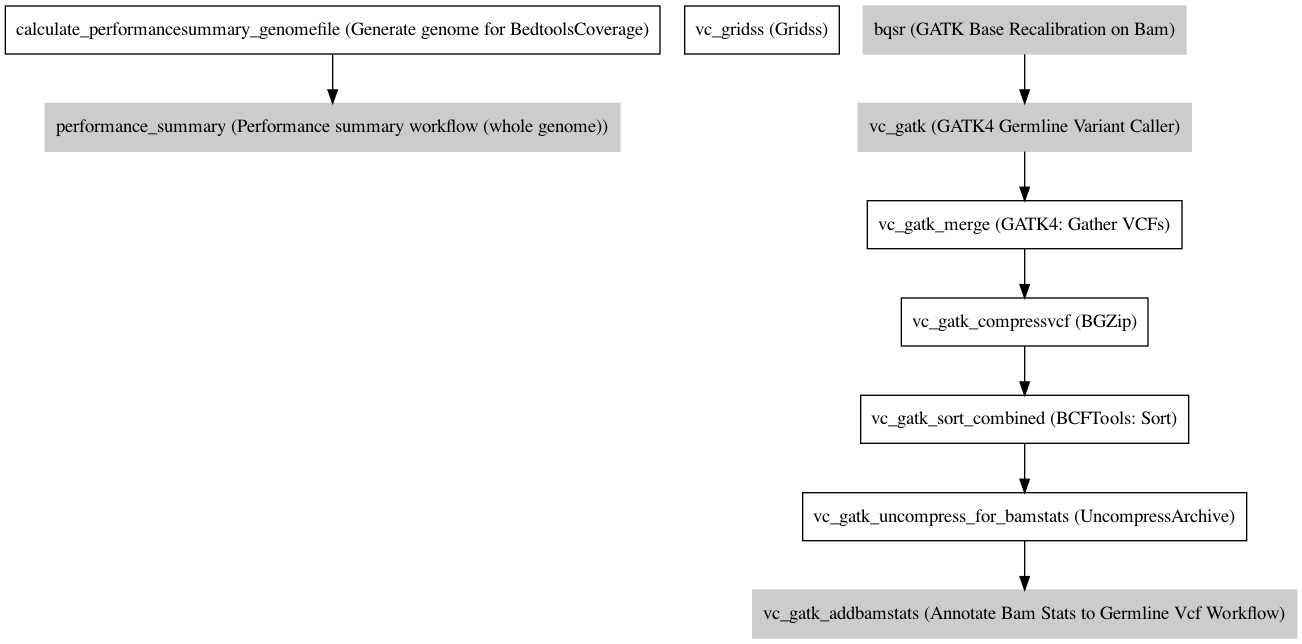
Information¶
| ID: | WGSGermlineGATKVariantsOnly |
|---|---|
| Versions: | 1.4.0 |
| Authors: | Michael Franklin, Richard Lupat, Jiaan Yu |
| Citations: | |
| Created: | 2018-12-24 |
| Updated: | 2020-06-22 |
Embedded Tools¶
| Generate genome for BedtoolsCoverage | GenerateGenomeFileForBedtoolsCoverage/v0.1.0 |
| Performance summary workflow (whole genome) | PerformanceSummaryGenome/v0.1.0 |
| Gridss | gridss/v2.6.2 |
| GATK Base Recalibration on Bam | GATKBaseRecalBQSRWorkflow/4.1.3 |
| GATK4 Germline Variant Caller | GATK4_GermlineVariantCaller/4.1.3.0 |
| GATK4: Gather VCFs | Gatk4GatherVcfs/4.1.3.0 |
| BGZip | bgzip/1.2.1 |
| BCFTools: Sort | bcftoolssort/v1.9 |
| UncompressArchive | UncompressArchive/v1.0.0 |
| Annotate Bam Stats to Germline Vcf Workflow | AddBamStatsGermline/v0.1.0 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| sample_name | String | Sample name from which to generate the readGroupHeaderLine for BwaMem |
| bam | IndexedBam | Input indexed bam (+ .bam.bai) to process. You only specify the primary sample.bam, and the index (eg: NA12878.bam.bai) will be picked up automatically. |
| reference | FastaWithIndexes | The reference genome from which to align the reads. This requires a number indexes (can be generated with the ‘IndexFasta’ pipeline This pipeline has been tested using the HG38 reference set.
|
| snps_dbsnp | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| snps_1000gp | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/ |
| known_indels | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| mills_indels | Gzipped<VCF> | From the GATK resource bundle, passed to BaseRecalibrator as known_sites |
| gridss_blacklist | bed | BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist |
| gatk_intervals | Array<bed> | List of intervals over which to split the GATK variant calling |
Workflow Description Language¶
version development
import "tools/GenerateGenomeFileForBedtoolsCoverage_v0_1_0.wdl" as G
import "tools/PerformanceSummaryGenome_v0_1_0.wdl" as P
import "tools/gridss_v2_6_2.wdl" as G2
import "tools/GATKBaseRecalBQSRWorkflow_4_1_3.wdl" as G3
import "tools/GATK4_GermlineVariantCaller_4_1_3_0.wdl" as G4
import "tools/Gatk4GatherVcfs_4_1_3_0.wdl" as G5
import "tools/bgzip_1_2_1.wdl" as B
import "tools/bcftoolssort_v1_9.wdl" as B2
import "tools/UncompressArchive_v1_0_0.wdl" as U
import "tools/AddBamStatsGermline_v0_1_0.wdl" as A
workflow WGSGermlineGATKVariantsOnly {
input {
String sample_name
File bam
File bam_bai
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
File snps_dbsnp
File snps_dbsnp_tbi
File snps_1000gp
File snps_1000gp_tbi
File known_indels
File known_indels_tbi
File mills_indels
File mills_indels_tbi
File gridss_blacklist
Array[File] gatk_intervals
}
call G.GenerateGenomeFileForBedtoolsCoverage as calculate_performancesummary_genomefile {
input:
reference=reference,
reference_dict=reference_dict
}
call P.PerformanceSummaryGenome as performance_summary {
input:
bam=bam,
bam_bai=bam_bai,
sample_name=sample_name,
genome_file=calculate_performancesummary_genomefile.out
}
call G2.gridss as vc_gridss {
input:
bams=[bam],
bams_bai=[bam_bai],
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
blacklist=gridss_blacklist
}
scatter (g in gatk_intervals) {
call G3.GATKBaseRecalBQSRWorkflow as bqsr {
input:
bam=bam,
bam_bai=bam_bai,
intervals=g,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi,
snps_1000gp=snps_1000gp,
snps_1000gp_tbi=snps_1000gp_tbi,
known_indels=known_indels,
known_indels_tbi=known_indels_tbi,
mills_indels=mills_indels,
mills_indels_tbi=mills_indels_tbi
}
}
scatter (Q in zip(gatk_intervals, transpose([bqsr.out, bqsr.out_bai]))) {
call G4.GATK4_GermlineVariantCaller as vc_gatk {
input:
bam=Q.right[0],
bam_bai=Q.right[1],
intervals=Q.left,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict,
snps_dbsnp=snps_dbsnp,
snps_dbsnp_tbi=snps_dbsnp_tbi
}
}
call G5.Gatk4GatherVcfs as vc_gatk_merge {
input:
vcfs=vc_gatk.out
}
call B.bgzip as vc_gatk_compressvcf {
input:
file=vc_gatk_merge.out
}
call B2.bcftoolssort as vc_gatk_sort_combined {
input:
vcf=vc_gatk_compressvcf.out
}
call U.UncompressArchive as vc_gatk_uncompress_for_bamstats {
input:
file=vc_gatk_sort_combined.out
}
call A.AddBamStatsGermline as vc_gatk_addbamstats {
input:
bam=bam,
bam_bai=bam_bai,
vcf=vc_gatk_uncompress_for_bamstats.out,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
output {
File out_performance_summary = performance_summary.performanceSummaryOut
File out_gridss_assembly = vc_gridss.assembly
File out_variants_gridss = vc_gridss.out
File out_variants = vc_gatk_sort_combined.out
Array[File] out_variants_split = vc_gatk.out
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: WGS Germline (GATK) [VARIANTS only]
doc: |
This is a genomics pipeline to ONLY call variants using GATK and GRIDSS from an indexed bam. The final variants are outputted in the VCF format.
This workflow is a reference pipeline using the Janis Python framework (pipelines assistant).
- Call variants using GRIDSS and GATK4;
- Outputs the final variants in the VCF format.
**Resources**
This pipeline has been tested using the HG38 reference set, available on Google Cloud Storage through:
- https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
This pipeline expects the assembly references to be as they appear in that storage (".fai", ".amb", ".ann", ".bwt", ".pac", ".sa", "^.dict").
The known sites (snps_dbsnp, snps_1000gp, known_indels, mills_indels) should be gzipped and tabix indexed.
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
- class: ScatterFeatureRequirement
- class: SubworkflowFeatureRequirement
- class: MultipleInputFeatureRequirement
inputs:
- id: sample_name
doc: Sample name from which to generate the readGroupHeaderLine for BwaMem
type: string
- id: bam
doc: |-
Input indexed bam (+ .bam.bai) to process. You only specify the primary sample.bam, and the index (eg: NA12878.bam.bai) will be picked up automatically.
type: File
secondaryFiles:
- pattern: .bai
- id: reference
doc: |2-
The reference genome from which to align the reads. This requires a number indexes (can be generated with the 'IndexFasta' pipeline This pipeline has been tested using the HG38 reference set.
This pipeline expects the assembly references to be as they appear in the GCP example. For example:
- HG38: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
- (".fai", ".amb", ".ann", ".bwt", ".pac", ".sa", "^.dict").
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: snps_dbsnp
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: snps_1000gp
doc: |-
From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``. Accessible from the HG38 genomics-public-data google cloud bucket: https://console.cloud.google.com/storage/browser/genomics-public-data/references/hg38/v0/
type: File
secondaryFiles:
- pattern: .tbi
- id: known_indels
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: mills_indels
doc: From the GATK resource bundle, passed to BaseRecalibrator as ``known_sites``
type: File
secondaryFiles:
- pattern: .tbi
- id: gridss_blacklist
doc: |-
BED file containing regions to ignore. For more information, visit: https://github.com/PapenfussLab/gridss#blacklist
type: File
- id: gatk_intervals
doc: List of intervals over which to split the GATK variant calling
type:
type: array
items: File
outputs:
- id: out_performance_summary
doc: A text file of performance summary of bam
type: File
outputSource: performance_summary/performanceSummaryOut
- id: out_gridss_assembly
doc: Assembly returned by GRIDSS
type: File
outputSource: vc_gridss/assembly
- id: out_variants_gridss
doc: Variants from the GRIDSS variant caller
type: File
outputSource: vc_gridss/out
- id: out_variants
doc: Merged variants from the GATK caller
type: File
outputSource: vc_gatk_sort_combined/out
- id: out_variants_split
doc: Unmerged variants from the GATK caller (by interval)
type:
type: array
items: File
outputSource: vc_gatk/out
steps:
- id: calculate_performancesummary_genomefile
label: Generate genome for BedtoolsCoverage
in:
- id: reference
source: reference
run: tools/GenerateGenomeFileForBedtoolsCoverage_v0_1_0.cwl
out:
- id: out
- id: performance_summary
label: Performance summary workflow (whole genome)
in:
- id: bam
source: bam
- id: sample_name
source: sample_name
- id: genome_file
source: calculate_performancesummary_genomefile/out
run: tools/PerformanceSummaryGenome_v0_1_0.cwl
out:
- id: performanceSummaryOut
- id: vc_gridss
label: Gridss
in:
- id: bams
source:
- bam
linkMerge: merge_nested
- id: reference
source: reference
- id: blacklist
source: gridss_blacklist
run: tools/gridss_v2_6_2.cwl
out:
- id: out
- id: assembly
- id: bqsr
label: GATK Base Recalibration on Bam
doc: Perform base quality score recalibration
in:
- id: bam
source: bam
- id: intervals
source: gatk_intervals
- id: reference
source: reference
- id: snps_dbsnp
source: snps_dbsnp
- id: snps_1000gp
source: snps_1000gp
- id: known_indels
source: known_indels
- id: mills_indels
source: mills_indels
scatter:
- intervals
run: tools/GATKBaseRecalBQSRWorkflow_4_1_3.cwl
out:
- id: out
- id: vc_gatk
label: GATK4 Germline Variant Caller
in:
- id: bam
source: bqsr/out
- id: intervals
source: gatk_intervals
- id: reference
source: reference
- id: snps_dbsnp
source: snps_dbsnp
scatter:
- intervals
- bam
scatterMethod: dotproduct
run: tools/GATK4_GermlineVariantCaller_4_1_3_0.cwl
out:
- id: variants
- id: out_bam
- id: out
- id: vc_gatk_merge
label: 'GATK4: Gather VCFs'
in:
- id: vcfs
source: vc_gatk/out
run: tools/Gatk4GatherVcfs_4_1_3_0.cwl
out:
- id: out
- id: vc_gatk_compressvcf
label: BGZip
in:
- id: file
source: vc_gatk_merge/out
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: vc_gatk_sort_combined
label: 'BCFTools: Sort'
in:
- id: vcf
source: vc_gatk_compressvcf/out
run: tools/bcftoolssort_v1_9.cwl
out:
- id: out
- id: vc_gatk_uncompress_for_bamstats
label: UncompressArchive
in:
- id: file
source: vc_gatk_sort_combined/out
run: tools/UncompressArchive_v1_0_0.cwl
out:
- id: out
- id: vc_gatk_addbamstats
label: Annotate Bam Stats to Germline Vcf Workflow
in:
- id: bam
source: bam
- id: vcf
source: vc_gatk_uncompress_for_bamstats/out
- id: reference
source: reference
run: tools/AddBamStatsGermline_v0_1_0.cwl
out:
- id: out
id: WGSGermlineGATKVariantsOnly