Tutorial 2: CWL Workflow -> Nextflow¶
Sections¶
- Introduction
- Running Janis Translate
- Translation Output
- Running the Translated Workflow
- Manual Adjustments
- Errors
- Completed Workflow
- Conclusion
Introduction¶
This section demonstrates translation of a CWL Workflow to Nextflow using janis translate.
The workflow used in this tutorial is taken from the McDonnell Genome Institute (MGI) analysis-workflows repository.
This resource stores publically available analysis pipelines for genomics data.
It is a fantastic piece of research software, and the authors thank MGI for their contribution to open-source research software.
The workflow using in this tutorial - align_sort_markdup.cwl - accepts multiple unaligned readsets as input and produces a single polished alignment bam file.
Main Inputs
- Unaligned reads stored across multiple BAM files
- Reference genome index
Main Outputs
- Single BAM file storing alignment for all readsets
Steps
- Read alignment (run in parallel across all readsets) -
bwa mem - Merging alignment BAM files to single file -
samtools merge - Sorting merged BAM by coordinate -
sambamba sort - Tagging duplicate reads in alignment -
picard MarkDuplicates - Indexing final BAM -
samtools index
Software
Before continuing, ensure you have the following software installed:
IDE
Any IDE or a CLI text editor (VIM, nano) are sufficient for this material.
We recommend Visual Studio Code (VS Code) as it is lightweight and has rich support for extensions to add functionality.
Obtaining Janis
In this tutorial we will use a singularity container to run janis translate.
Containers are great because they remove the need for package managers, and guarantee that the software can run on any machine.
Run the following command to pull the janis image:
singularity pull janis.sif docker://pppjanistranslate/janis-translate:0.13.0
Check your image by running the following command:
singularity exec ~/janis.sif janis translate
If the image is working, you should see the janis translate helptext.
Downloading Source Files and Sample Data
For this tutorial we will fetch all necessary data from zenodo using wget.
This archive contains the source CWL file, sample data, and finished translations as a reference.
Run the following commands to download & decompress the zenodo archive:
wget https://zenodo.org/record/8275567/files/tutorial2.tar.gz
tar -xvf tutorial2.tar.gz
After you have decompressed the tar archive, change into the new directory:
cd tutorial2
Inside this folder we have the following structure:
tutorial2
├── data
│ ├── 2895499223.bam
│ ├── 2895499237.bam
│ ├── chr17_test.dict
│ ├── chr17_test.fa
│ ├── chr17_test.fa.amb
│ ├── chr17_test.fa.ann
│ ├── chr17_test.fa.bwt
│ ├── chr17_test.fa.fai
│ ├── chr17_test.fa.pac
│ └── chr17_test.fa.sa
├── final
│ ├── main.nf
│ ├── modules
│ │ ├── align_and_tag.nf
│ │ ├── index_bam.nf
│ │ ├── mark_duplicates_and_sort.nf
│ │ ├── merge_bams_samtools.nf
│ │ └── name_sort.nf
│ ├── nextflow.config
│ ├── subworkflows
│ │ └── align.nf
│ └── templates
│ └── markduplicates_helper.sh
└── source
├── align_sort_markdup.cwl
├── subworkflows
│ └── align.cwl
└── tools
├── align_and_tag.cwl
├── index_bam.cwl
├── mark_duplicates_and_sort.cwl
├── merge_bams_samtools.cwl
└── name_sort.cwl
We will translate the source/align_sort_markdup.cwl workflow into nextflow using janis, then will test out our translation using the files inside the indexed bam file in the data/ folder.
Running Janis Translate¶
To translate a tool / workflow, we use janis translate.
janis translate --from <src> --to <dest> <filepath>
The --from argument specifies the workflow language of the source file(s), and --to specifies the destination we want to translate to.
In our case, this will be --from cwl --to nextflow.
The <filepath> argument is the source file we will translate.
In this tutorial, the filepath will be source/align_sort_markdup.cwl
To translate our cwl tool, run the following command:
singularity exec ~/janis.sif janis translate --from cwl --to nextflow source/align_sort_markdup.cwl
You will see a folder called translated/ appear, with various nextflow files and directories inside.
Translation Output¶
The output translation will contain multiple files and directories.
You will see a folder called translated/ appear - inside this folder, we should see the following structure:
translated
├── main.nf # main workflow (align_sort_markdup)
├── modules # folder containing nextflow processes
│ ├── align_and_tag.nf
│ ├── index_bam.nf
│ ├── mark_duplicates_and_sort.nf
│ ├── merge_bams_samtools.nf
│ └── name_sort.nf
├── nextflow.config # config file to supply input information
├── subworkflows # folder containing nextflow subworkflows
│ └── align.nf
└── templates # folder containing any scripts used by processes
└── markduplicates_helper.sh
Now that we have performed translation using janis translate, we need to check the translated workflow for correctness.
From here, we will do a test-run of the workflow using sample data, and make manual adjustments to the translated workflow where needed.
Running the Translated Workflow¶
Inspect main.nf
The main workflow translation appears as main.nf in the translated/ folder.
This filename is just a convention, and we use it to provide clarity about the main entry point of the workflow.
In our case main.nf is equivalent to the main CWL Workflow file we translated: align_sort_markdup.cwl.
NOTE:
Before continuing, feel free to have a look at the other nextflow files which have been generated during translation:
Each CWL Subworkflow appears as a nextflowworkflowin thesubworkflows/directory.
Each CWL CommandLineTool appears as a nextflowprocessin themodules/directory.
In main.nf we see the nextflow workflows / processes called by the main workflow:
include { ALIGN } from './subworkflows/align'
include { INDEX_BAM } from './modules/index_bam'
include { MARK_DUPLICATES_AND_SORT } from './modules/mark_duplicates_and_sort'
include { MERGE_BAMS_SAMTOOLS as MERGE } from './modules/merge_bams_samtools'
include { NAME_SORT } from './modules/name_sort'
We also see that some nextflow Channels and a single variable have been set up.
These are used to supply data according to nextflow’s adoption of the dataflow programming model.
// data which will be passed as channels
ch_bams = Channel.fromPath( params.bams ).toList()
ch_readgroups = Channel.of( params.readgroups ).toList()
// data which will be passed as variables
reference = params.reference.collect{ file(it) }
Focusing on the channel declarations, we want to note a few things:
ch_bamsis analygous to the ‘bams’ input inalign_sort_markdup.cwl.
It declares a queue channel which expects the data supplied viaparams.bamsarepathtypes.
It then groups the bams together as a sole emission using.toList().
We will need to set upparams.bamsto supply this data.ch_readgroupsis analygous to the ‘readgroups’ input inalign_sort_markdup.cwl.
It is the same asch_bams, except it requiresvaltypes rather thanpathtypes.
We will need to set upparams.readgroupsto supply this data.
We also see a new variable called reference being created.
This reference var is analygous to the ‘reference’ input in align_sort_markdup.cwl.
It collects the ‘reference’ primary & secondary files together in a list.
We will need to set up params.reference to supply this data.
Note: Why doesreferenceappear as a variable, rather than aChannel?
In Nextflow, we find it is best to create Channels for data that moves through the pipeline.
Thech_bamschannel is created because we want to consume / transform the input bams during the pipeline.
On the other hand, thereferencegenome variable is not something we really want to consume or transform.
It’s more like static helper data which can be used by processes, but it doesn’t move through our pipeline, and definitely shouldn’t be consumed or transformed in any way.
Note:.toList()
Nextflow queue channels work differently to lists.
Instead of supplying all items together, queue channels emit each item separately.
This results in a separate task being spawned for each item in the queue when the channel is used.
As the CWL workflow input specifies thatbamsis a list, we use.toList()to group all items as a sole emission.
This mimics a CWL array which is the datatype of thebamsinputs.
As it turns out, the CWL workflow ends up running thealignstep in parallel across thebams&readgroupsinputs.
Parallelisation in nextflow happens by default.
To facilitate this, the.flatten()method is called onch_bamsandch_readgroupswhen used in theALIGNtask.
This emits items inch_bamsandch_readgroupsindividually, spawning a newALIGNtask for each pair.
We’re kinda doing redundant work by calling.toList(), then.flatten()whench_bamsandch_readgroupsare used.
janis translateisn’t smart enough yet to detect this yet, but may do so in future.
Continuing down main.nf, we see the main workflow {} has 5 tasks.
Each task has been supplied values according to the source workflow. Comments display the name of the process/workflow input which is being fed a particular value.
workflow {
ALIGN(
ch_bams.flatten(), // bam
reference, // reference
ch_readgroups.flatten() // readgroup
)
INDEX_BAM(
MARK_DUPLICATES_AND_SORT.out.sorted_bam.map{ tuple -> tuple[0] } // bam
)
MARK_DUPLICATES_AND_SORT(
params.mark_duplicates_and_sort.script, // script
NAME_SORT.out.name_sorted_bam, // bam
params.NULL_VALUE, // input_sort_order
params.final_name // output_name
)
MERGE(
ALIGN.out.tagged_bam.toList(), // bams
params.final_name // name
)
NAME_SORT(
MERGE.out.merged_bam // bam
)
}
Before main.nf can be run, we will need to supply values for the params variables.
This is done in nextflow.config.
Inspect nextflow.config
To test the translated workflow, we will first set up workflow inputs in nextflow.config.
Before running a workflow, nextflow will attempt to open nextflow.config and read in config information and global param variables from this file.
We use this file to tell nextflow how to run and to supply workflow inputs.
Inside the translated/ folder you will see that nextflow.config is already provided.
Janis translate creates this file to provide clarity about the necessary workflow inputs, and to set some other config variables.
Open nextflow.config and have a look at the contents. It should look similar to the following:
nextflow.enable.dsl = 2
singularity.enabled = true
singularity.autoMounts = true
singularity.cacheDir = "$HOME/.singularity/cache"
params {
// Placeholder for null values.
// Do not alter unless you know what you are doing.
NULL_VALUE = 'NULL'
// WORKFLOW OUTPUT DIRECTORY
outdir = './outputs'
// INPUTS (MANDATORY)
bams = [] // (MANDATORY array) eg. [file1, ...]
reference = [] // (MANDATORY fastawithindexes) eg. [fasta, amb, ann, bwt, dict, fai, pac, sa]
readgroups = NULL_VALUE // (MANDATORY array) eg. [string1, ...]
// INPUTS (OPTIONAL)
final_name = "final.bam"
// PROCESS: ALIGN_AND_TAG
align_and_tag.cpus = 8
align_and_tag.memory = 20000
// PROCESS: INDEX_BAM
index_bam.memory = 4000
// PROCESS: MARK_DUPLICATES_AND_SORT
mark_duplicates_and_sort.script = "/home2/training/align_sort_markdup/templates/markduplicates_helper.sh"
mark_duplicates_and_sort.cpus = 8
mark_duplicates_and_sort.memory = 40000
// PROCESS: MERGE_BAMS_SAMTOOLS
merge_bams_samtools.cpus = 4
merge_bams_samtools.memory = 8000
// PROCESS: NAME_SORT
name_sort.cpus = 8
name_sort.memory = 26000
}
NOTE:NULL_VALUE = 'NULL'
Nextflow doesn’t likenullvalues to be passed to process inputs.
This is a challenge for translation as other languages allowoptionalinputs.
To get around this, Janis Translate sets theparams.NULL_VALUEvariable as anullplaceholder forvaltype inputs.
You will see this being used in nextflow processes to do optionality checking.
The auto-generated nextflow.config splits up workflow inputs using some headings.
// INPUTS (MANDATORY)
Workflow inputs which are required to run the workflow. We must provide a value for these.
// INPUTS (OPTIONAL)
Workflow inputs which are not required to run the workflow. These are optional.
// PROCESS: ALIGN_AND_TAG
Inputs which are specific to a particular process. These are usually static values rather than files.May be mandatory or optional.
Setting up Workflow Inputs
Janis Translate will enter values for workflow inputs where possible. Others need to be manually supplied as they are specific to the input data you wish to use.
In our case, we need to supply values for those under the // INPUTS (MANDATORY) heading.
Specifically, we need to provide sample data for the bams, reference, and readgroups inputs.
Copy and paste the following text, to override the params under the // INPUTS (MANDATORY) heading:
// INPUTS (MANDATORY)
bams = [
"../data/2895499223.bam",
"../data/2895499237.bam",
]
reference = [
"../data/chr17_test.fa",
"../data/chr17_test.fa.amb",
"../data/chr17_test.fa.ann",
"../data/chr17_test.fa.bwt",
"../data/chr17_test.fa.fai",
"../data/chr17_test.dict",
"../data/chr17_test.fa.pac",
"../data/chr17_test.fa.sa",
]
readgroups = [
'@RG\tID:2895499223\tPU:H7HY2CCXX.3.ATCACGGT\tSM:H_NJ-HCC1395-HCC1395\tLB:H_NJ-HCC1395-HCC1395-lg24-lib1\tPL:Illumina\tCN:WUGSC',
'@RG\tID:2895499237\tPU:H7HY2CCXX.4.ATCACGGT\tSM:H_NJ-HCC1395-HCC1395\tLB:H_NJ-HCC1395-HCC1395-lg24-lib1\tPL:Illumina\tCN:WUGSC'
]
Run the Workflow
Ensure you are in the translated/ working directory, where nextflow.config and main.nf reside.
If not, use the following to change directory.
cd translated
To run the workflow using our sample data, we can now write the following command:
nextflow run main.nf
While the workflow runs, you will encounter this error:
Access to 'MARK_DUPLICATES_AND_SORT.out' is undefined since the process 'MARK_DUPLICATES_AND_SORT' has not been invoked before accessing the output attribute
This is somewhat expected. Janis translate doesn’t produce perfect translations - just the best it can do.
This is the first of 3 errors we will encounter and fix while making this workflow runnable.
Manual Adjustments¶
Translations performed by janis translate often require manual changes due to the difficulty of translating between languages with non-overlapping feature sets.
In this section we will fix 3 errors to bring the translation to a finished state.
NOTE
If having trouble during this section, the finished workflow is available in thetutorial2/finalfolder for reference.
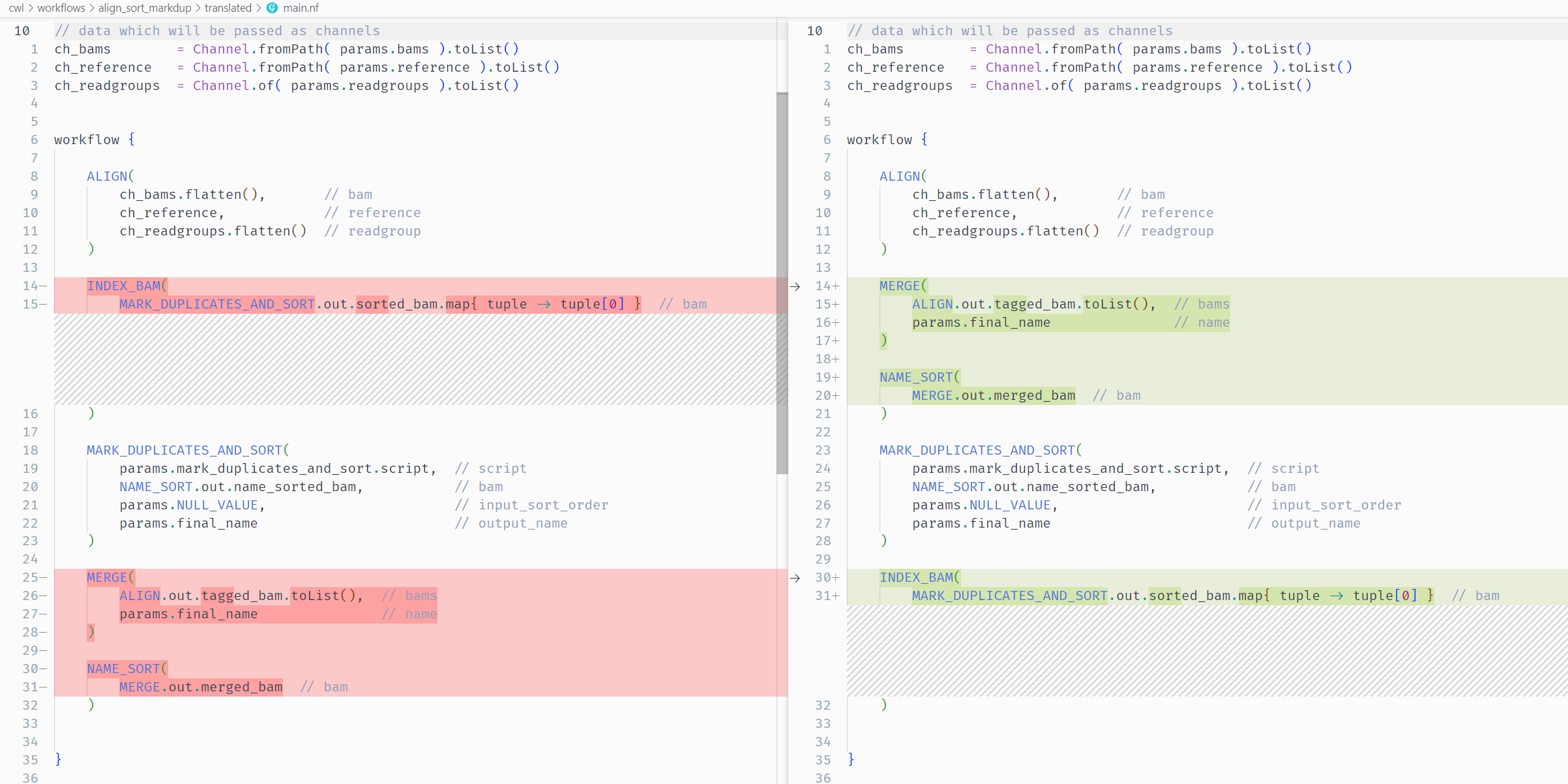
Error 1: Process Order¶
Error message
The first issue we need to address is caused by tasks being in the wrong order.
Access to 'MARK_DUPLICATES_AND_SORT.out' is undefined since the process 'MARK_DUPLICATES_AND_SORT' has not been invoked before accessing the output attribute
This nextflow error message is quite informative, and tells us that a task is trying to access the output of MARK_DUPLICATES_AND_SORT before it has run.
The offending task is INDEX_BAM as it uses MARK_DUPLICATES_AND_SORT.out.sorted_bam.map{ tuple -> tuple[0] } as an input value to the process.
NOTE
Why does this happen?
Internally, CWL stores a list of tasks to execute alphabetically.
It then works out the required order of operations at runtime.
This is different to nextflow, where the order of tasks in the nextflow file is their execution order.
Sincejanis translatecurrently usescwltoolto load cwl documents, janis is passed the alphabetical list of tasks from CWL. This may or may not be the correct order when translated to nextflow!
Troubleshooting
It seems that a nextflow process is trying to run before a prior process has finished.
This is most likely due to the processes in our main.nf workflow being in the wrong order.
Let’s look at the source CWL align_sort_markdup.cwl workflow to view the correct order of steps:
steps:
align:
...
merge:
...
name_sort:
...
mark_duplicates_and_sort:
...
index_bam:
...
This differs from our translated main.nf which has the following order:
workflow {
ALIGN(
...
)
INDEX_BAM(
...
)
MARK_DUPLICATES_AND_SORT(
...
)
MERGE(
...
)
NAME_SORT(
...
)
}
Solution
We will need to rearrange the process calls in main.nf so they mirror the order seen in the source CWL.
TASK ⚡
Rerranging process calls
- Cut-and-paste the process calls to be in the correct order in the main workflow.
Show Correct Order
1. ALIGN 2. MERGE 3. NAME_SORT 4. MARK_DUPLICATES_AND_SORT 5. INDEX_BAMShow Changes (main.nf)
workflow { ALIGN( ch_bams.flatten(), // ch_bam reference, // ch_reference ch_readgroups.flatten() // ch_readgroup ) MERGE( ALIGN.out.tagged_bam.toList(), // bams params.final_name // name ) NAME_SORT( MERGE.out.merged_bam // bam ) MARK_DUPLICATES_AND_SORT( params.mark_duplicates_and_sort.script, // script NAME_SORT.out.name_sorted_bam, // bam params.NULL_VALUE, // input_sort_order params.final_name // output_name ) INDEX_BAM( MARK_DUPLICATES_AND_SORT.out.sorted_bam.map{ tuple -> tuple[0] } // bam ) }
After you are done, rerun the workflow by using the same command as before.
nextflow run main.nf
Error 2: Unquoted Strings¶
Error message
The second error is due to the readgroup input of the ALIGN_AND_TAG process being used without enclosing quotes.
Caused by:
Process `ALIGN:ALIGN_AND_TAG (1)` terminated with an error exit status (1)
Command executed:
/bin/bash /usr/bin/alignment_helper.sh 2895499223.bam @RG INCLID:2895499223RY_PU:H7HY2CCXX.3.ATCACGGTITY=ISM:H_NJ-HCC1395-HCC1395ILB:H_NJ-HCC1395-HCC1395-lg24-lib1LEVEL=5PL:IlluminaSCN:WUGSC chr17_test.fa 8 > refAlign.bam
The issue can be seen in the command above.
We see /bin/bash /usr/bin/alignment_helper.sh 2895499223.bam which is expected.
This is then followed by what looks like 2 string arguments: @RG and INCLID....
This is causing the problem.
Troubleshooting
The Command executed section in nextflow error messages is particularly useful.
This message is printed to the shell, but can also be seen by navigating to the process working directory & viewing .command.sh.
Let’s look at the nextflow ALIGN_AND_TAG process so we can match up which process input is causing the error.
Indiviual arguments have been marked with their (position) to aid our investigation.
process ALIGN_AND_TAG {
container "mgibio/alignment_helper-cwl:1.0.0"
publishDir "${params.outdir}/align_and_tag"
cpus "${params.align_and_tag.cpus}"
memory "${params.align_and_tag.memory}"
input:
path reference
path bam
val readgroup
output:
path "refAlign.bam", emit: aligned_bam
script:
def reference = reference[0]
"""
(1) /bin/bash (2) /usr/bin/alignment_helper.sh \
(3) ${bam} \
(4) ${readgroup} \
(5) ${reference} \
(6) 8 \
(7) > (8) refAlign.bam
"""
}
Here is the command executed with the same numbering:
(1) /bin/bash (2) /usr/bin/alignment_helper.sh (3) 2895499223.bam (4) @RG (5) ID:2895499223 PU:H7HY2CCXX.3.ATCACGGT SM:H_NJ-HCC1395-HCC1395 LB:H_NJ-HCC1395-HCC1395-lg24-lib1 PL:Illumina CN:WUGSC (6) chr17_test.fa (7) 8 (8) > (9) refAlign.bam
Matching up the two, we can see that arguments (1-3) match their expected values.
Argument (4) starts out right, as we expect the readgroup input.
Looking in the script: section of the nextflow process, we expect the ${reference} input to appear as argument (5), but in the actual command it appears as argument (6) chr17_test.fa.
The issue seems to be that the readgroup input has been split into 2 strings, instead of 1 single string.
By tracing back through the workflow, we can track that params.readgroups supplies the value for readgroup in this nextflow process:
modules/align_and_tag.nf: readgroup
subworkflows/align.nf: ch_readgroup
main.nf: ch_readgroups.flatten()
main.nf: ch_readgroups = Channel.of( params.readgroups ).toList()
Looking at nextflow.config we see that this particular readgroup value is supplied as follows:
@RG\tID:2895499223\tPU:H7HY2CCXX.3.ATCACGGT\tSM:H_NJ-HCC1395-HCC1395\tLB:H_NJ-HCC1395-HCC1395-lg24-lib1\tPL:Illumina\tCN:WUGSC
The @RG being split from the rest of the readgroup value, and the text which follows is incorrect in the command.
The issue here is that this value in params.readgroups contains spaces and tabs (\t).
When used in a nextflow process, string arugments should be enclosed using "".
Solution
Back in the modules/align_and_tag.nf file, let’s properly enclose the readgroup input in quotes.
TASK ⚡
Enclosing
readgroupin quotes
- In the
ALIGN_AND_TAGprocess script enclose the${readgroup}reference in quotes.Show Changes (align_and_tag.nf)
process ALIGN_AND_TAG { ... script: def reference = reference[0] """ /bin/bash /usr/bin/alignment_helper.sh \ ${bam} \ "${readgroup}" \ <- quotes added ${reference} \ 8 \ > refAlign.bam """ } ...
After you are have made the change, re-run the workflow by using the same command as before:
nextflow run main.nf
Error 3: Filename Clashes¶
Error Message
The final error is flagged by the nextflow workflow engine.
Upon re-running the workflow, you will encounter the following message:
Process `MERGE` input file name collision -- There are multiple input files for each of the following file names: refAlign.bam
This informs us that more than 1 file with the name refAlign.bam have been inputs to MERGE process.
Nextflow does not allow this behaviour.
Other workflow engines use temporary names for files so that name clashes are impossible.
Nextflow requires us to be more specific with our filenames so we can track which file is which.
This is not a hard-and-fast rule of workflow engines, but nextflow enforces using unique names to encourage best-practises.
Troubleshooting
To track the cause, let’s look at the data which feeds the MERGE process.
main.nf
MERGE(
ALIGN.out.tagged_bam.toList(), // bams
params.final_name // name
)
We can see the translated nextflow workflow is collecting the tagged_bam output of all ALIGN tasks as a list using .toList().
This value feeds the bams input of the MERGE process.
The issue is that 2+ files in this list must have the same filename: “refAlign.bam”.
We need to see how files end up being placed into ALIGN.out.tagged_bam to track where the error is occuring.
Looking in the ALIGN subworkflow in subworkflows/align.nf, we see the offending tagged_bam output emit.
This gets its data from the aligned_bam output of the ALIGN_AND_TAG process in modules/align_and_tag.nf.
The source of the issue must be that the aligned_bam output is producing the same filename each time the task is run.
Opening modules/align_and_tag.nf we can see how the align_bam output is created & collected:
output:
path "refAlign.bam", emit: aligned_bam <-
script:
def reference = reference[0]
"""
/bin/bash /usr/bin/alignment_helper.sh \
${bam} \
"${readgroup}" \
${reference} \
8 \
> refAlign.bam <-
"""
From looking at the output and script section of this process, we can see that the aligned_bam output will always have the same filename.
In the script, we generate a file called refAlign.bam by redirecting stdout to this file.
In the output, refAlign.bam is collected as the output.
This would be fine if our workflow was supplied only a single BAM input file, but we want our workflow to run when we have many BAMs.
Solution
To fix this issue, we need to give the output files unique names.
This can be accomplished using a combination of the path bam process input, and the .simpleName operator.
As our input BAM files will all have unique names, we can use their name as a base for our output alignemnts. If we were to use their filename directly, we would get their file extension too, which we don’t want!
Luckily the .simpleName nextflow operator will return the basename of a file without directory path or extensions.
For example, imagine we have a process input path reads.
Suppose it gets fed the value "data/SRR1234.fq" at runtime.
Calling ${reads.simpleName} in the process script would yield "SRR1234".
Note that directory path and the extension have been trimmed out!
TASK ⚡
Unique output names for the
ALIGN_AND_TAGprocess
- In the script section, use
.simpleNameto generate an output filename based on thepath bamprocess input.
Add “_refAlign.bam” onto the end of this filename for clarity.- Alter the output collection expression for the
aligned_bamoutput to collect this file.Show Changes (align_and_tag.nf)
process ALIGN_AND_TAG { container "mgibio/alignment_helper-cwl:1.0.0" publishDir "${params.outdir}/align_and_tag" cpus "${params.align_and_tag.cpus}" memory "${params.align_and_tag.memory}" input: path reference path bam val readgroup output: path "${bam.simpleName}_refAlign.bam", emit: aligned_bam <- script: def reference = reference[0] """ /bin/bash /usr/bin/alignment_helper.sh \ ${bam} \ "${readgroup}" \ ${reference} \ 8 \ > ${bam.simpleName}_refAlign.bam <- """ }
After you are have made these changes, re-run the workflow by using the same command as before:
nextflow run main.nf
With any luck, this will fix the remaining issues and the workflow will now run to completion.
Completed Workflow¶
Once completed, we can check the outputs/ folder to view our results.
If everything went well, the outputs/ folder should have the following structure:
outputs
├── align_and_tag
│ ├── 2895499223_refAlign.bam
│ └── 2895499237_refAlign.bam
├── index_bam
│ ├── final.bam
│ └── final.bam.bai
├── mark_duplicates_and_sort
│ ├── final.bam
│ ├── final.bam.bai
│ └── final.mark_dups_metrics.txt
├── merge_bams_samtools
│ └── final.bam.merged.bam
└── name_sort
└── final.NameSorted.bam
Conclusion¶
In this section we explored how to translate the align_sort_markdup CWL Workflow to Nextflow using janis translate.
If having trouble, the finished workflow is also available in the tutorial2/final folder for reference.
In addition, the following diffs shows the changes we made to files during manual adjustment of the workflow.
main.nf
 alt text
alt text
align_and_tag.nf
 alt text
alt text